
The Spatial Pattern of the Two Genetic Lineages of the Field Vole in Lithuania


 ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction, Polymerase Chain Reaction (PCR) and Sequencing
2.3. Data Analysis
3. Results
3.1. Genetic Variation of M. agrestis
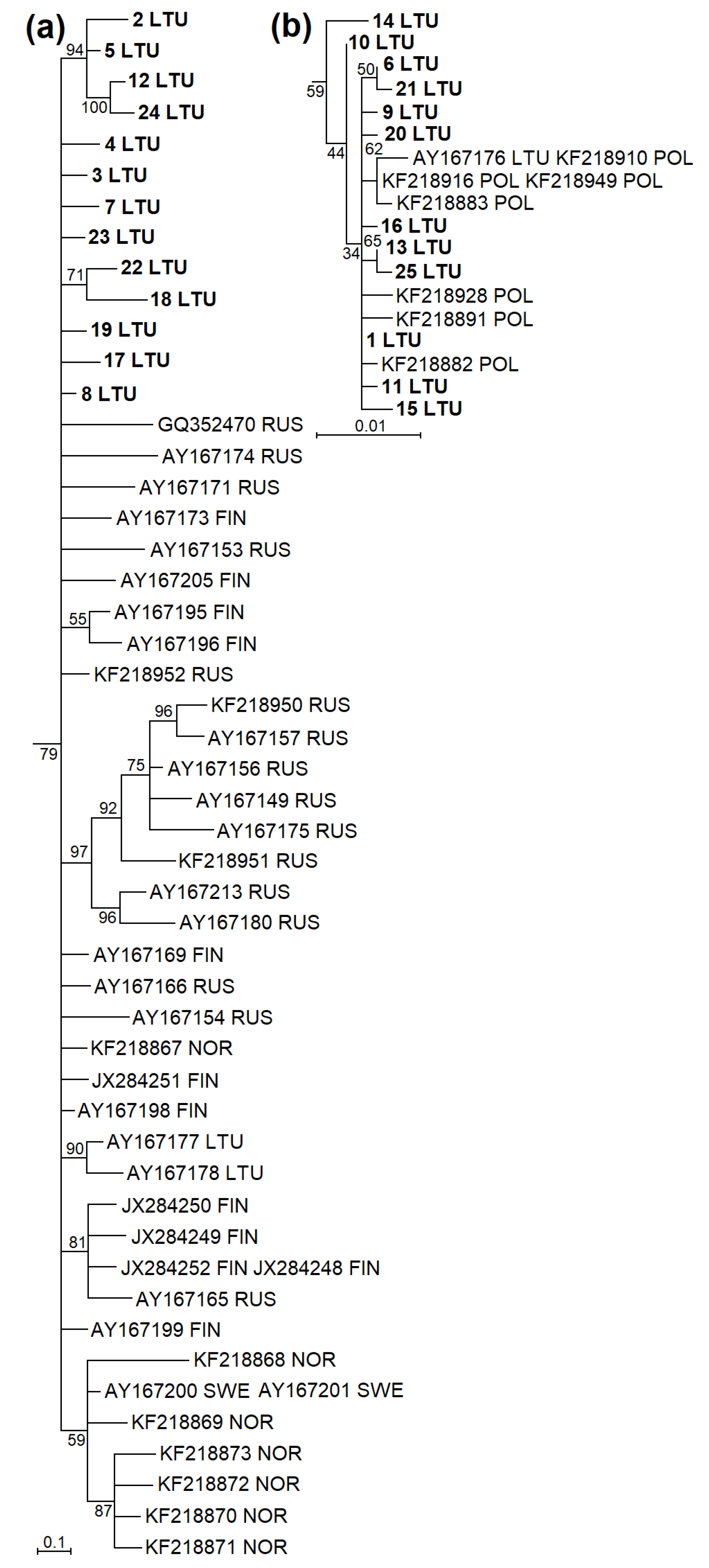
3.2. Phylogenetic Groups of M. agrestis
3.3. Genetic Comparison of Lithuanian M. agrestis Populations
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kryštufek, B.; Vohralík, V.; Zima, J.; Zagorodnyuk, I. Microtus agrestis (Errata Version Published in 2017). The IUCN Red List of Threatened Species 2016: E.T13426A115112050. 2016. Available online: https://doi.org/10.2305/IUCN.UK.2016-3.RLTS.T13426A22349665.en (accessed on 6 September 2022).
- Shenbrot, G.I.; Krasnov, B.R. An Atlas of the Geographic Distribution of the Arvicoline Rodents of the World (Rodentia, Muridae: Arvicolinae); Pensoft Publishers: Sofia, Bulgaria, 2005; 336p. [Google Scholar]
- Mathias, M.D.L.; Hart, E.B.; Ramalhinho, M.D.G.; Jaarola, M. Microtus agrestis (Rodentia: Cricetidae). Mamm. Species 2017, 49, 23–39. [Google Scholar] [CrossRef]
- Gromov, I.M.; Polyakov, I.Y. Fauna of the USSR, Vol. 3 Voles (Microtinae); Brill Publishing Co.: Leiden, The Netherlands, 1992; pp. 297–483. [Google Scholar]
- Zima, J. Microtus agrestis. In The Atlas of European Mammals; Mitchell-Jones, A.J., Amori, G., Bogdanowicz, W., Kryštufek, B., Reijnders, P.J.H., Spitzenberger, F., Stubbe, M., Thissen, J.B.M., Vohralík, V., Zima, J., Eds.; Academic Press: London, UK, 1999; pp. 225–227. [Google Scholar]
- Balčiauskas, L.; Trakimas, G.; Juškaitis, R.; Ulevičius, A.; Balčiauskienė, L. Atlas of Lithuanian Mammals, Amphibians and Reptiles, 2nd ed.; Akstis: Vilnius, Lithuania, 1999; p. 112. [Google Scholar]
- Stirkė, V.; Balčiauskas, L.; Balčiauskienė, L. Common Vole as a Focal Small Mammal Species in Orchards of the Northern Zone. Diversity 2021, 13, 134. [Google Scholar] [CrossRef]
- Balčiauskas, L.; Balčiauskienė, L.; Garbaras, A.; Stirkė, V. Diversity and Diet Differences of Small Mammals in Commensal Habitats. Diversity 2021, 13, 346. [Google Scholar] [CrossRef]
- Kitrytė, N. Diversity of Small Mammal Parasites and Factors Shaping Their Communities. Ph.D. Dissertation, Nature Research Centre, Vilnius University, Vilnius, Lithuania, 2022. [Google Scholar]
- Jaarola, M.; Tegelström, H. Colonization history of north European field voles (Microtus agrestis) revealed by mitochondrial DNA. Mol. Ecol. 1995, 4, 299–310. [Google Scholar] [CrossRef]
- Jaarola, M.; Searle, J.B. Phylogeography of field voles (Microtus agrestis) in Eurasia inferred from mitochondrial DNA sequences. Mol. Ecol. 2002, 11, 2613–2621. [Google Scholar] [CrossRef]
- Baltrūnaitė, L.; Mažeikytė, R.; Stunžėnas, V. New Data on the Distribution of Mitochondrial DNA Lineages of the Fiels Vole (Microtus agrestis) in Lithuania and Belarus. Acta Zool. Litu. 2006, 16, 115–118. [Google Scholar] [CrossRef]
- Stojak, J.; Wójcik, J.; Ruczyńska, I.; Searle, J.B.; McDevitt, A.D. Contrasting and congruent patterns of genetic structuring in two Microtus vole species using museum specimens. Mamm. Res. 2016, 61, 141–152. [Google Scholar] [CrossRef]
- Stojak, J.; Borowik, T.; Górny, M.; McDevitt, A.D.; Wójcik, J.M. Climatic influences on the genetic structure and distributiion of the common vole and field vole in Europe. Mamm. Res. 2019, 64, 19–29. [Google Scholar] [CrossRef]
- Ivanter, E.V.; Kurkhinen, Y.P.; Sokolov, A.V. Ecology of the field vole (Microtus agrestis L.) in indigenous and anthropogenic landscapes of eastern Fennoscandia. Russ. J. Ecol. 2013, 44, 213–220. [Google Scholar] [CrossRef]
- Stirkė, V.; Balčiauskas, L.; Balčiauskienė, L. Spatiotemporal Variation of Small Mammal Communities in Commercial Orchards across the Small Country. Agriculture 2022, 12, 632. [Google Scholar] [CrossRef]
- Prūsaitė, J. Lithuanian Fauna. Mammals; Mokslas: Vilnius, Lietuva, 1988; p. 295. [Google Scholar]
- Aljanabi, S.M.; Martinez, I. Universal and Rapid Salt-extraction of High Quality Genomic DNA for PCR-based Techniques. Nucleic Acids Res. 1997, 25, 4692–4693. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new Capabilities and Interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Villesen, P. FaBox: An Online Toolbox for Fasta Sequences. Mol. Ecol. Notes 2007, 7, 965–968. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Bandelt, H.-J.; Forster, P.; Röhl, A. Median-Joining Networks for Inferring Intraspecific Phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (Version 3.0): An Integrated Software Package for Population Genetics Data Analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Nei, M. Genetic Distance between Populations. Am. Nat. 1972, 106, 283–292. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic Analysis in Excel. Population Genetic Software for Teaching and Research—An Update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Patwardhan, A.; Ray, S.; Roy, A. Molecular Markers in Phylogenetic Studies—A Review. J. Phylogen. Evolution. Biol. 2014, 2, 131. [Google Scholar] [CrossRef]
- Tobe, S.S.; Kitchener, A.C.; Linacre, A.M.T. Reconstructing Mammalian Phylogenies: A Detailed Comparison of the Cytochrome b and Cytochrome Oxidase Subunit I Mitochondrial Genes. PLoS ONE 2010, 5, e14156. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, M.; Ali, H.S.; Stieger, N.; Groschup, M.H.; Wolf, R.; Ulrich, R.G. Molecular Identification of Small Mammal Species Using Novel Cytochrome b Gene-Derived Degenerated Primers. Biochem. Genet. 2011, 50, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, A.; Darvish, J.; Aliabadian, M.; Yazdani Moghaddam, F.; Kryštufek, B. New insight into the cradle of the grey voles (subgenus Microtus) inferred from mitochondrial cytochrome b sequences. Mammalia 2017, 81, 583. [Google Scholar] [CrossRef]
- Nygren, J.; Rasmuson, M. Allozyme variation in natural populations of field vole (Microtus agrestis L.) I. Survey of the “Semi-stable” population in southern Sweden. Hereditas 1980, 92, 65–72. [Google Scholar] [CrossRef]
- Jaarola, M.; Tegelström, H.; Fredga, K. A contact zone with non-coincident clines for sex-specific markers in the field vole (Microtus agrestis). Evolution 1997, 51, 241–249. [Google Scholar]
- Jaarola, M.; Tegelström, H.; Fredga, K. Colonization history in Fennoscandian rodents. Biol. J. Linn. Soc. 1999, 68, 113–127. [Google Scholar] [CrossRef]
- Jaarola, M.; Martínková, N.; Gündüz, İ.; Brunhoff, C.; Zima, J.; Nadachowski, A.; Amori, G.; Bulatova, N.S.; Chondropoulos, B.; Fraguedakis-Tsolis, S.; et al. Molecular phylogeny of the speciose vole genus Microtus (Arvicolinae, Rodentia) inferred from mitochondrial DNA sequences. Mol. Phylogenet. Evol. 2004, 33, 647–663. [Google Scholar] [CrossRef]
- Mammal Species of the World. A Taxonomic and Geographic Reference, 3rd ed.; Wilson, D.E., Reeder, D.M., Eds.; Johns Hopkins University Press: Baltimore, US, 2005; Volume 2, pp. 956–1038. [Google Scholar]
- Handbook of the Mammals of the World, Rodents II; Wilson, D.E., Mittermeier, R.A., Lacher, T.E., Eds.; Lynx Edicions: Barcelona, Spain, 2017; Volume 7, pp. 333–334. [Google Scholar]
- Cheprakov, M.I.; Chernousova, N.F. Variability of the Ecological and Morphological Characteristics of Field Vole (Microtus agrestis) in the Southern and Middle Urals. Russ. J. Ecol. 2020, 51, 565–569. [Google Scholar] [CrossRef]
- Jiang, J.-Q.; Wu, S.-X.; Chen, J.-J.; Liu, C.-Z. Characterization of the complete mitochondrial genome of short-tailed field vole, Microtus agrestis. Mitochondr. DNA Part B 2018, 3, 845–846. [Google Scholar] [CrossRef]
- Jancewicz, E.; Falkowska, E.; Ratkiewicz, M. mtDNA evidence for a local northern latitude Pleistocene refugium for the root vole (Microtus oeconomus, Arvicolinae, Rodentia) from Eastern Poland. J. Zool. Syst. Evol. Res. 2015, 53, 331–339. [Google Scholar] [CrossRef]
- Stojak, J.; McDevitt, A.D.; Herman, J.S.; Kryštufek, B.; Uhlíková, J.; Purger, J.J.; Lavrenchenko, L.A.; Searle, J.B.; Wójcik, J.M. Between the Balkans and the Baltic: Phylogeography of a Common Vole Mitochondrial DNA Lineage Limited to Central Europe. PLoS ONE 2016, 11, e0168621. [Google Scholar] [CrossRef] [PubMed]
- Czarnomska, S.D.; Niedziałkowska, M.; Borowik, T.; Jędrzejewska, B. Regional and local patterns of genetic variation and structure in yellow-necked mice—The roles of geographic distance, population abundance, and winter severity. Ecol. Evol. 2018, 8, 8171–8186. [Google Scholar] [CrossRef] [PubMed]
- Łopucki, R.; Mróz, I.; Nowak-Życzyńska, Z.; Perlińska-Teresiak, M.; Owadowska-Cornil, E.; Klich, D. Genetic Structure of the Root Vole Microtus oeconomus: Resistance of the Habitat Specialist to the Natural Fragmentation of Preferred Moist Habitats. Genes 2022, 13, 434. [Google Scholar] [CrossRef] [PubMed]
- Balčiauskas, L.; Alejūnas, P. Small mammal species diversity and abundance in Žagarė Regional Park. Acta Zool. Litu. 2011, 21, 163–172. [Google Scholar] [CrossRef]
- Čepukienė, A.; Jasiulionis, M. Small mammal community changes during forest succession (Pakruojis district, north Lithuania). Zool. Ecol. 2012, 22, 144–149. [Google Scholar] [CrossRef]
- Balčiauskas, L.; Čepukienė, A.; Balčiauskienė, L. Small mammal community response to early meadow–forest succession. Forest Ecosystems 2017, 4, 11. [Google Scholar] [CrossRef]
- Balčiauskas, L.; Skipitytė, R.; Balčiauskienė, L.; Jasiulionis, M. Resource partitioning confirmed by isotopic signatures allows small mammals to share seasonally flooded meadows. Ecol. Evol. 2019, 9, 5479–5489. [Google Scholar] [CrossRef]
- Beysard, M.; Perrin, N.; Jaarola, M.; Heckel, G.; Vogel, P. Asymmetric and differential gene introgression at a contact zone between two highly divergent lineages of field voles (Microtus agrestis). J. Evolution. Biol. 2012, 25, 400–408. [Google Scholar] [CrossRef]
- Herman, J.S.; Searle, J.B. Post-glacial partitioning of mitochondrial genetic variation in the field vole. P. R. Soc. B. 2011, 278, 3601–3607. [Google Scholar] [CrossRef]
- Herman, J.S.; McDevitt, A.D.; Kawałko, A.; Jaarola, M.; Wójcik, J.M.; Searle, J.B. Land-Bridge Calibration of Molecular Clocks and the Post-Glacial Colonization of Scandinavia by the Eurasian Field Vole Microtus agrestis. PLoS ONE 2014, 9, e103949. [Google Scholar] [CrossRef]
- Myers, P.; Lundrigan, B.L.; Hoffman, S.M.G.; Haraminac, A.P.; Seto, S.H. Climate-induced changes in the small mammal communities of the Northern Great Lakes Region. Glob. Chang. Biol. 2009, 15, 1434–1454. [Google Scholar] [CrossRef]
- Mitchell, D.; Snelling, E.P.; Hetem, R.S.; Maloney, S.K.; Strauss, W.M.; Fuller, A. Revisiting concepts of thermal physiology: Predicting responses of mammals to climate change. J. Anim. Ecol. 2018, 87, 956–973. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, M.; Visconti, P.; Rondinini, C. A framework for the identification of hotspots of climate change risk for mammals. Glob. Change Biol. 2018, 24, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Zárybnická, M.; Riegert, J.; Bejček, V.; Sedláček, F.; Šťastný, K.; Šindelář, J.; Heroldová, M.; Vilímová, J.; Zima, J. Long-term changes of small mammal communities in heterogenous landscapes of Central Europe. Eur. J. Wildl. Res. 2017, 63, 89. [Google Scholar] [CrossRef]
- Fuentes-Montemayor, E.; Ferryman, M.; Watts, K.; Macgregor, N.A.; Hambly, N.; Brennan, S.; Coxon, R.; Langridge, H.; Park, K.J. Small mammal responses to long-term large-scale woodland creation: The influence of local and landscape-level attributes. Ecol. Appl. 2019, 30, e02028. [Google Scholar] [CrossRef]
- Marcinkevičiūtė, L.; Vilkevičiūtė, J.; Žukovskis, J.; Pranskūnienė, R. Social Dimensions of Projected Climate Change Impacts on Ecosystem Services in the Coastal-Rural Area of Nemunas River Reaches and Curonian Lagoon (Lithuania). Water 2021, 13, 1114. [Google Scholar] [CrossRef]
- Tripolskaja, L.; Kazlauskaitė-Jadzevičė, A. Trend Analyses of Percolation of Atmospheric Precipitation Due to Climate Change: Case Study in Lithuania. Agronomy 2022, 12, 1784. [Google Scholar] [CrossRef]
- Senetra, A.; Szczepańska, A.; Veteikis, D.; Wasilewicz-Pszczółkowska, M.; Šimanauskienė, R.; Volungevičius, J. Changes of the land use patterns in the Polish and Lithuanian trans-border rural area. Baltica 2013, 26, 157–168. [Google Scholar] [CrossRef]
- Veteikis, D.; Piškinaitė, E. Geografiniai žemėnaudos kaitos tyrimai Lietuvoje: Raida, kryptys, perspektyvos. Geologija. Geografija 2019, 5, 14–29. [Google Scholar] [CrossRef]
- Mizaras, S.; Doftartė, A.; Lukminė, D.; Šilingienė, R. Sustainability of Small-Scale Forestry and Its Influencing Factors in Lithuania. Forests 2020, 11, 619. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | GenBank acc. No. | N | Sequence Similarity (%) |
|---|---|---|---|
| Belarus | AY167155 | 3 | 98.76–99.59 |
| Finland | AY167169, AY167173, AY167195-96, AY167198-99, AY167205, JX284248-51 | 13 | 98.49–99.73 |
| Romania | AY167172 | 3 | 98.49–99.04 |
| Lithuania | AY167176-78 | 3 | 98.35–99.86 |
| Denmark | AY167179, AY167184, AY167194, GU563297-99, MT410884 | 8 | 98.21–99.31 |
| Czechia | AY167151-52, AY167212 | 4 | 98.07–99.59 |
| Germany | AY167210, DQ662095-102, DQ768138-42, DQ768145-47, GU563292-96, MK535087-92 | 28 | 98.07–99.59 |
| Sweden | AY167167-68, AY167200-201, AY167203-204, AY167206-209, AY167211 | 12 | 98.07–99.59 |
| Russia | AY167149, AY167153-54, AY16715356-57, AY167165-66, AY167171, AY167174-75, AY167180, AY167213, KF218950-52, GQ352470 | 16 | 97.94–99.59 |
| Scotland | AY167181-82, AY167190, FJ619746-52, FJ619754-FJ619758, FJ619760-63, GU563195-239 | 105 | 97.94–99.59 |
| Luxembourg | GU563291 | 1 | 97.94–98.76 |
| Netherlands | AY167183 | 1 | 97.94–98.76 |
| Poland | AY167176, AY167185, DQ662095, KF218875-949 | 109 | 97.80–99.86 |
| Norway | AY167202, KF218851-73 | 29 | 97.66–99.59 |
| Wales | FJ619778, FJ619782, FJ619784-86, GU563260-67 | 17 | 97.52–99.18 |
| England | AY167150, AY167170, AY167191-93, AY167197, FJ619753, FJ619759, FJ619764-77, FJ619779-81, FJ619783, GU563240-59, GU563268-83 | 105 | 97.25–99.45 |
| France | AY167188-89, GU563284-90, JX284281-83 | 15 | 92.85–99.59 |
| Switzerland | AY167158-61, MW478038 | 5 | 92.85–99.04 |
| Spain | AY167162-64, AY167187, JX284265, JX284273-80 | 30 | 91.88–94.22 |
| Portugal | AY167186, JX284253-64, JX284266-72 | 67 | 91.61–93.26 |
| Sample | n | h | S | K | Hd ± SD | π ± SD |
|---|---|---|---|---|---|---|
| cytb | ||||||
| Switzerland | 5 | 4 | 47 | 27.700000 | 1.000 ± 0.126 | 0.03810 ± 0.01049 |
| France | 15 | 11 | 59 | 19.35238 | 0.943 ± 0.045 | 0.02662 ± 0.00669 |
| Russia | 16 | 16 | 43 | 8.24167 | 1.000 ± 0.022 | 0.01134 ± 0.00104 |
| Poland | 109 | 66 | 72 | 7.38473 | 0.989 ± 0.003 | 0.01016 ± 0.00042 |
| Spain | 30 | 13 | 50 | 7.37471 | 0.903 ± 0.031 | 0.01014 ± 0.00234 |
| Czechia | 4 | 3 | 12 | 7.16667 | 0.833 ± 0.222 | 0.00986 ± 0.00258 |
| Norway | 29 | 24 | 44 | 7.05419 | 0.985 ± 0.014 | 0.00970 ± 0.00115 |
| Germany | 28 | 13 | 21 | 6.30072 | 0.920 ± 0.038 | 0.00867 ± 0.00052 |
| England | 105 | 52 | 56 | 6.20180 | 0.875 ± 0.029 | 0.00853 ± 0.00063 |
| Sweden | 12 | 10 | 22 | 5.87879 | 0.970 ± 0.044 | 0.00809 ± 0.00114 |
| Wales | 17 | 10 | 25 | 4.97059 | 0.919 ± 0.044 | 0.00684 ± 0.00110 |
| Scotland | 105 | 52 | 61 | 4.97289 | 0.957 ± 0.010 | 0.00684 ± 0.00033 |
| Finland | 13 | 11 | 16 | 3.92308 | 0.974 ± 0.039 | 0.00540 ± 0.00072 |
| Denmark | 8 | 7 | 13 | 3.60714 | 0.964 ± 0.077 | 0.00496 ± 0.00203 |
| Portugal | 67 | 12 | 15 | 1.70240 | 0.767 ± 0.046 | 0.00234 ± 0.00032 |
| Joniškis | 29 | 11 | 19 | 5.38916 | 0.845 ± 0.045 | 0.00741 ± 0.00038 |
| Pakruojis | 24 | 11 | 17 | 4.63768 | 0.851 ± 0.054 | 0.00638 ± 0.00066 |
| Zarasai | 8 | 4 | 11 | 4.39286 | 0.821 ± 0.101 | 0.00604 ± 0.00138 |
| Rusnė | 13 | 2 | 2 | 0.30769 | 0.154 ± 0.126 | 0.00042 ± 0.00035 |
| Lithuania * | 77 | 28 | 35 | 5.04272 | 0.841 ± 0.038 | 0.00694 ± 0.00039 |
| Overall | 648 | 331 | 240 | 20.98218 | 0.990 ± 0.001 | 0.02890 ± 0.00001 |
| D-loop | ||||||
| Pakruojis | 24 | 8 | 16 | 4.59058 | 0.819 ± 0.050 | 0.01093 ± 0.00172 |
| Joniškis | 29 | 9 | 12 | 4.52422 | 0.829 ± 0.047 | 0.01077 ± 0.00061 |
| Zarasai | 8 | 3 | 9 | 3.50000 | 0.607 ± 0.164 | 0.00833 ± 0.00278 |
| Rusnė | 13 | 3 | 4 | 0.74359 | 0.295 ± 0.156 | 0.00177 ± 0.00097 |
| Overall | 74 | 19 | 25 | 4.81599 | 0.890 ± 0.021 | 0.01147 ± 0.00070 |
| Joniškis | Pakruojis | Zarasai | Rusnė | |
|---|---|---|---|---|
| Joniškis | 0.17332 ** | 0.06341 | 0.35922 *** | |
| Pakruojis | 0.08884 * | 0.31469 * | 0.11994 * | |
| Zarasai | 0.06349 | 0.16033 * | 0.67878 *** | |
| Rusnė | 0.34939 *** | 0.23161 ** | 0.68462 *** |
| Site | Average, 2004–2020 | In the Year of Genetic Sampling | |||
|---|---|---|---|---|---|
| % | RA | % | RA | Source | |
| Joniškis | 3.3 | 0.67 | 2.2 | 0.49 | [43] |
| Pakruojis | 9.1 | 1.11 | 12.6 | 1.00 | [44] |
| Zarasai | 7.9 | 0.11 | 3.1 | 0.12 | [45] |
| Rusnė | 1.5 | 0.27 | 7.6 | 0.70 | [46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prakas, P.; Butkauskas, D.; Vaišvilienė, J.; Balčiauskienė, L.; Balčiauskas, L. The Spatial Pattern of the Two Genetic Lineages of the Field Vole in Lithuania. Diversity 2022, 14, 820. https://doi.org/10.3390/d14100820
Prakas P, Butkauskas D, Vaišvilienė J, Balčiauskienė L, Balčiauskas L. The Spatial Pattern of the Two Genetic Lineages of the Field Vole in Lithuania. Diversity. 2022; 14(10):820. https://doi.org/10.3390/d14100820
Chicago/Turabian StylePrakas, Petras, Dalius Butkauskas, Jevgenija Vaišvilienė, Laima Balčiauskienė, and Linas Balčiauskas. 2022. "The Spatial Pattern of the Two Genetic Lineages of the Field Vole in Lithuania" Diversity 14, no. 10: 820. https://doi.org/10.3390/d14100820
APA StylePrakas, P., Butkauskas, D., Vaišvilienė, J., Balčiauskienė, L., & Balčiauskas, L. (2022). The Spatial Pattern of the Two Genetic Lineages of the Field Vole in Lithuania. Diversity, 14(10), 820. https://doi.org/10.3390/d14100820