
Genetic Structure and Diversity of the Yellowbelly Threadfin Bream Nemipterus bathybius in the Northern South China Sea

 , , and
, , and
Abstract
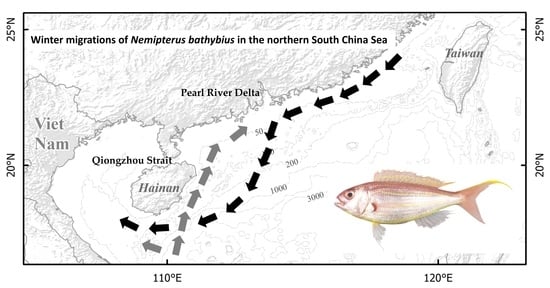
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Sequencing
2.2. Diversity and Structure Analyses
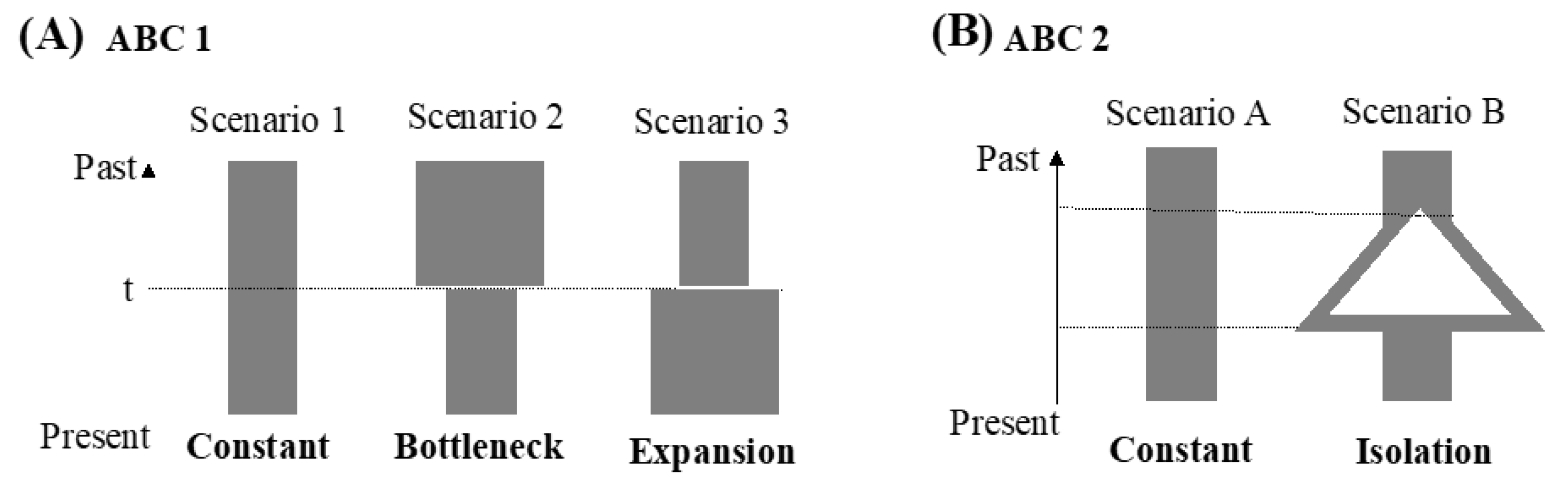
2.3. Demographic Analyses
3. Results
3.1. Genetic Diversity
3.2. Population Differentiation
3.3. Demography
4. Discussion
4.1. Genetic Structure and Diversity
4.2. Demographic History
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kizaki, K.; Oshiro, I. The origin of the Ryukyu Islands. In Natural History of the Ryukyus; Tsukiji Shokan: Tokyo, Japan, 1980; pp. 8–37. (In Japanese) [Google Scholar]
- Ota, H. Biogeographical characteristics of terrestrial vertebrates of the Ryukyu Archioelago. In Search of Origins of Okinawan Culture; Bunshinlnsatsu: Naha, Japan, 1994; pp. 460–464. (In Japanese) [Google Scholar]
- Voris, H.K. Maps of Pleistocene sea levels in Southeast Asia: Shorelines, river systems and time durations. J. Biogeogr. 2000, 27, 1153–1167. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.-N.; Jamandre, B.W.; Hsu, C.-C.; Tzeng, W.-N.; Durand, J.-D. Plio-Pleistocene sea level and temperature fluctuations in the northwestern Pacific promoted speciation in the globally-distributed flathead mullet Mugil cephalus. BMC Evol. Biol. 2011, 11, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ni, G.; Li, Q.; Kong, L.; Zheng, X. Phylogeography of Bivalve Cyclina sinensis: Testing the Historical Glaciations and Changjiang River Outflow Hypotheses in Northwestern Pacific. PLoS ONE 2012, 7, e49487. [Google Scholar] [CrossRef] [Green Version]
- Ni, G.; Li, Q.; Kong, L.; Yu, H. Comparative phylogeography in marginal seas of the northwestern Pacific. Mol. Ecol. 2014, 23, 534–548. [Google Scholar] [CrossRef]
- He, L.; Mukai, T.; Chu, K.H.; Ma, Q.; Zhang, J. Biogeographical role of the Kuroshio Current in the amphibious mudskipper Periophthalmus modestus indicated by mitochondrial DNA data. Sci. Rep. 2015, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.-W.; Wang, H.-S.; Han, G.-D.; Ke, C.-H.; Zhan, X.; Nakano, T.; Williams, G.A. The Impact of Yangtze River Discharge, Ocean Currents and Historical Events on the Biogeographic Pattern of Cellana toreuma along the China Coast. PLoS ONE 2012, 7, e36178. [Google Scholar] [CrossRef] [Green Version]
- Ni, G.; Kern, E.; Dong, Y.; Li, Q.; Park, J.-K. More than meets the eye: The barrier effect of the Yangtze River outflow. Mol. Ecol. 2017, 26, 4591–4602. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.-D.; Wang, X.-L.; Zhan, R. Nutrient conditions in the Yellow Sea and the East China Sea. Estuar. Coast. Shelf Sci. 2003, 58, 127–136. [Google Scholar] [CrossRef]
- Liu, J.-X.; Gao, T.-X.; Wu, S.-F.; Zhang, Y.-P. Pleistocene isolation in the Northwestern Pacific marginal seas and limited dispersal in a marine fish, Chelon haematocheilus (Temminck & Schlegel, 1845). Mol. Ecol. 2007, 16, 275–288. [Google Scholar] [CrossRef]
- Chen, C.-C.; Shiah, F.-K.; Chiang, K.-P.; Gong, G.-C.; Kemp, W.M. Effects of the Changjiang (Yangtze) River discharge on planktonic community respiration in the East China Sea. J. Geophys. Res. Space Phys. 2009, 114. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Chan, T.-Y.; Tsang, L.M.; Chu, K.H. Phylogeography of the mitten crab Eriocheir sensu stricto in East Asia: Pleistocene isolation, population expansion and secondary contact. Mol. Phylogenetics Evol. 2009, 52, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tsang, L.M.; Dong, Y.-W. Causations of phylogeographic barrier of some rocky shore species along the Chinese coastline. BMC Evol. Biol. 2015, 15, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Kong, L.; Chen, J.; Matsukuma, A.; Li, Q. Phylogeography of bivalve Meretrix petechialis in the Northwestern Pacific indicated by mitochondrial and nuclear DNA data. PLoS ONE 2017, 12, e0183221. [Google Scholar] [CrossRef] [Green Version]
- Qu, M.; Tang, W.; Liu, Q.; Wang, D.; Ding, S. Genetic diversity within grouper species and a method for interspecific hybrid identification using DNA barcoding and RYR3 marker. Mol. Phylogenetics Evol. 2018, 121, 46–51. [Google Scholar] [CrossRef]
- Maas, D.L.; Prost, S.; Bi, K.; Smith, L.L.; Armstrong, E.E.; Aji, L.P.; Toha, A.H.A.; Gillespie, R.G.; Becking, L.E. Rapid divergence of mussel populations despite incomplete barriers to dispersal. Mol. Ecol. 2018, 27, 1556–1571. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.E.; Sage, G.K.; Wedemeyer, K.; Sonsthagen, S.A.; Menning, D.M.; Gravley, M.C.; Sexson, M.G.; Nelson, R.J.; Talbot, S.L. Micro-geographic population genetic structure within Arctic cod (Boreogadus saida) in Beaufort Sea of Alaska. ICES J. Mar. Sci. 2019, 76, 1713–1721. [Google Scholar] [CrossRef]
- Peng, X.; Wu, X.; Zhang, Y.-P.; Zhang, H.; Tang, J.; Kang, B. Genetic diversity and metapopulation structures of two intertidal species along the coast of Zhejiang, China: Implications for conservation. Mitochondrial DNA Part A 2019, 30, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, G. Small-scale population genetic structure of the sand bubbler crab Scopimera ryukyuensis in the Ryukyu Islands, Japan. Mol. Biol. Rep. 2020, 47, 2619–2626. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Y. Status of Marine Biodiversity of the China Seas. PLoS ONE 2013, 8, e50719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.X. Response of western Pacific marginal seas to glacial cycles: Paleocenographic and sedimentological features. Mar. Geol. 1999, 156, 5–39. [Google Scholar] [CrossRef]
- Russell, B.C. FAO Species Catalogue, Vol 12. Nemipterid Fishes of the World. (Threadfin Breams, Whiptail Breams, Monocle Breams, Dwarf Monocle Breams, and Coral Breams). Family Nemipteridae. An Annotated and Illustrated Catalogue of Nemipterid Species Known to Date; FAO Fisheries Synopsis, no. 125, (VIII plates); FAO: Rome, Italy, 1990; 149p. [Google Scholar]
- Chen, Z.C.; Liu, J.X. Commercial Fishes of South China Sea; Guangdong Science and Technology Press: Guangzhou, China, 1982; pp. 184–188. (In Chinese) [Google Scholar]
- Granada, V.P.; Masuda, Y.; Matsuoka, T. Age and growth of the yellowbelly threadfin bream Nemipterus bathybius in Kagoshima Bay, southern Japan. Fish. Sci. 2004, 70, 497–506. [Google Scholar] [CrossRef]
- Chen, Z.-Z.; Kong, X.-L.; Xu, S.-N.; Qiu, Y.-S.; Huang, Z.-R. Dynamic changes of population parameters of Nemipterus bathybiusin the Beibu Gulf. J. Fish. China 2012, 36, 584–591. [Google Scholar] [CrossRef]
- Huang, Z.; Chen, Z. Stock actuality of Nemipterus bathybius in southwestern continental of Nansha Islands. J. Zhanjiang Ocean. Univ. 2005, 25, 35–38. [Google Scholar]
- Wu, R.; Niu, S.; Qin, C.; Liu, J. Complete mitochondrial genome of the yellowbelly threadfin bream, Nemipterus bathybius (Perciformes, Nemipteridae). Mitochondrial DNA Part A 2016, 27, 4624–4626. [Google Scholar] [CrossRef]
- Kong, X.; Chen, Z.; Lin, L.; Li, C.; Xu, S.; Liu, Y. Polymorphic microsatellite loci isolated from the yellowbelly threadfin bream, Nemipterus bathybius. Genet. Mol. Res. 2014, 13, 5254–5257. [Google Scholar] [CrossRef]
- Froese, R.; Pauly, D. FishBase. World Wide Web Electronic Publication. 2021. Available online: http://www.fishbase.org (accessed on 1 June 2021).
- Yang, J.-Q.; Hsu, K.-C.; Liu, Z.-Z.; Su, L.-W.; Kuo, P.-H.; Tang, W.-Q.; Zhou, Z.-C.; Liu, D.; Bao, B.-L.; Lin, H.-D. The population history of Garra orientalis (Teleostei: Cyprinidae) using mitochondrial DNA and microsatellite data with approximate Bayesian computation. BMC Evol. Biol. 2016, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Hsu, K.-C.; Luo, J.-Z.; Wang, C.-H.; Chan, B.-P.; Li, J.; Kuo, P.-H.; Lin, H.-D. Genetic diversity and population history ofTanichthys albonubes (Teleostei: Cyprinidae): Implications for conservation. Aquat. Conserv. Mar. Freshw. Ecosyst. 2018, 28, 422–434. [Google Scholar] [CrossRef]
- Han, C.-C.; Hsu, K.-C.; Fang, L.-S.; Cheng, I.-M.; Lin, H.-D. Geographical and temporal origins of Neocaridina species (Decapoda: Caridea: Atyidae) in Taiwan. BMC Genet. 2019, 20, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.-R.; Hsu, K.-C.; Yi, M.-R.; Li, B.; Wang, W.-K.; Kang, B.; Lin, H.-D. Cryptic diversity of the spotted scat Scatophagus argus (Perciformes: Scatophagidae) in the South China Sea: Pre- or post-production isolation. Mar. Freshw. Res. 2020, 71, 1640–1650. [Google Scholar] [CrossRef]
- Forcina, G.; Woutersen, K.; Sánchez-Ramírez, S.; Angelone, S.; Crampe, J.P.; Pérez, J.M.; Fandos, P.; Granados, J.E.; Jowers, M.J. Demography reveals populational expansion of a recently extinct Iberian ungulate. Zoosyst. Evol. 2021, 97, 211. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [Green Version]
- Nei, M.; Tajima, F. Maximum likelihood estimation of the number of nucleotide substitutions from restriction sites data. Genetetics 1983, 105, 207–217. [Google Scholar] [CrossRef]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [Green Version]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Dupanloup, I.; Schneider, S.; Excoffier, L. A simulated annealing approach to define the genetic structure of populations. Mol. Ecol. 2002, 11, 2571–2581. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.-X. Statistical Tests of Neutrality of Mutations Against Population Growth, Hitchhiking and Background Selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambau, A.; Suchard, M. BEAST 1.8.0. 2013. Available online: http://beast.bio.ed.ac.uk (accessed on 26 March 2021).
- Van Steenberge, M.W.; Vanhove, M.P.M.; Manda, A.C.; Larmuseau, M.H.D.; Swart, B.L.; Khang’Mate, F.; Arndt, A.; Hel-lemans, B.; Van Houdt, J.; Micha, J.; et al. Unravelling the evolution of Africa’s drainage basins through a widespread freshwater fish, the African sharptooth catfish Clarias gariepinus. J. Biogeogr. 2020, 47, 1739–1754. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Suchard, M. Tracer v1.6. 2013. Available online: http://tree.bio.ed.ac.uk/software/tracer/ (accessed on 1 May 2018).
- Cornuet, J.-M.; Pudlo, P.; Veyssier, J.; Dehne-Garcia, A.; Gautier, M.; Leblois, R.; Marin, J.-M.; Estoup, A. DIYABC v2.0: A software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 2014, 30, 1187–1189. [Google Scholar] [CrossRef] [Green Version]
- Pons, O.; Petit, R.J. Measwring and Testing Genetic Differentiation with Ordered Versus Unordered Alleles. Genetics 1996, 144, 1237–1245. [Google Scholar] [CrossRef]
- Ju, Y.-M.; Wu, J.-H.; Kuo, P.-H.; Hsu, K.-C.; Wang, W.-K.; Lin, F.-J.; Lin, H.-D. Mitochondrial genetic diversity of Rhinogobius giurinus (Teleostei: Gobiidae) in East Asia. Biochem. Syst. Ecol. 2016, 69, 60–66. [Google Scholar] [CrossRef]
- Qiu, F.; Li, H.; Lin, H.; Ding, S.; Miyamoto, M.M. Phylogeography of the inshore fish, Bostrychus sinensis, along the Pacific coastline of China. Mol. Phylogenetics Evol. 2016, 96, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Tsuda, Y.; Takayama, K.; Nagashima, R.; Tateishi, Y.; Kajita, T. The presence of a cryptic barrier in the West Pacific Ocean suggests the effect of glacial climate changes on a widespread sea-dispersed plant, Vigna marina (Fabaceae). Ecol. Evol. 2019, 9, 8429–8440. [Google Scholar] [CrossRef]
- Niu, S.-F.; Wu, R.-X.; Zhai, Y.; Zhang, H.; Li, Z.-L.; Liang, Z.-B.; Chen, Y.-H. Demographic history and population genetic analysis of Decapterus maruadsi from the northern South China Sea based on mitochondrial control region sequence. PeerJ 2019, 7, e7953. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.-C.; Ahmad, A.T.; Nuruddin, A.A.; Nor, S.A.M. Cytochromebgene reveals panmixia among Japanese Threadfin Bream, Nemipterus japonicus (Bloch, 1791) populations along the coasts of Peninsular Malaysia and provides evidence of a cryptic species. Mitochondrial DNA 2016, 27, 575–584. [Google Scholar] [CrossRef]
- Yang, J.; Wu, D.; Lin, X. On the dynamics of the South China Sea Warm Current. J. Geophys. Res. Space Phys. 2008, 113, 08003. [Google Scholar] [CrossRef] [Green Version]
- Ni, G.; Kim, T.; Shin, Y.; Park, J.; Lee, Y.; Kil, H.-J.; Park, J.-K. Life-history features and oceanography drive phylogeographic patterns of the chiton Acanthochitona cf. rubrolineata (Lischke, 1873) in the northwestern Pacific. PeerJ 2020, 8, e8794. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Li, Q.; Kong, L.; Yu, H. Cryptic diversity of marine gastropod Monodonta labio (Trochidae): Did the early Pleistocene glacial isolation and sea surface temperature gradient jointly drive diversification of sister species and/or subspecies in the Northwestern Pacific? Mar. Ecol. 2017, 38, e12443. [Google Scholar] [CrossRef]
- Wang, Z.-D.; Liao, J.; Huang, C.-Q.; Long, S.-S.; Zhang, S.; Guo, Y.-S.; Liu, L.; Liu, C.-W. Significant genetic differentiation of Gobiopterus lacustris, a newly recorded transparent goby in China. Mitochondrial DNA Part A 2018, 29, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Delrieu-Trottin, E.; Mona, S.; Maynard, J.; Neglia, V.; Veuille, M.; Planes, S. Population expansions dominate demographic histories of endemic and widespread Pacific reef fishes. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, T.L.; Castilho, R.; Stevens, J.R. Meta-analysis of northeast Atlantic marine taxa shows contrasting phylogeographic patterns following post-LGM expansions. PeerJ 2018, 6, e5684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Zhang, A.-B.; Weese, D.; Li, S.; Li, J.; Zhang, J. Demographic response of cutlassfish (Trichiurus japonicus and T. nanhaiensis) to fluctuating palaeo-climate and regional oceanographic conditions in the China seas. Sci. Rep. 2014, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Templeton, A.R. The ‘Eve’ Hypotheses: A Genetic Critique and Reanalysis. Am. Anthr. 1993, 95, 51–72. [Google Scholar] [CrossRef]
- Li, H.; Lin, H.; Li, J.; Ding, S. Phylogeography of the Chinese Beard Eel, Cirrhimuraena chinensis Kaup, Inferred from Mitochondrial DNA: A Range Expansion after the Last Glacial Maximum. Int. J. Mol. Sci. 2014, 15, 13564–13577. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Huang, Z.R.; Xu, Y.W.; Chen, Z.Z. Population genetic structure of brushtooth lizardfish (Saurida undosquamis) based om mitochondrial cytochrome b gene sequences. South China Fish. Sci. 2019, 15, 41–48. [Google Scholar] [CrossRef]
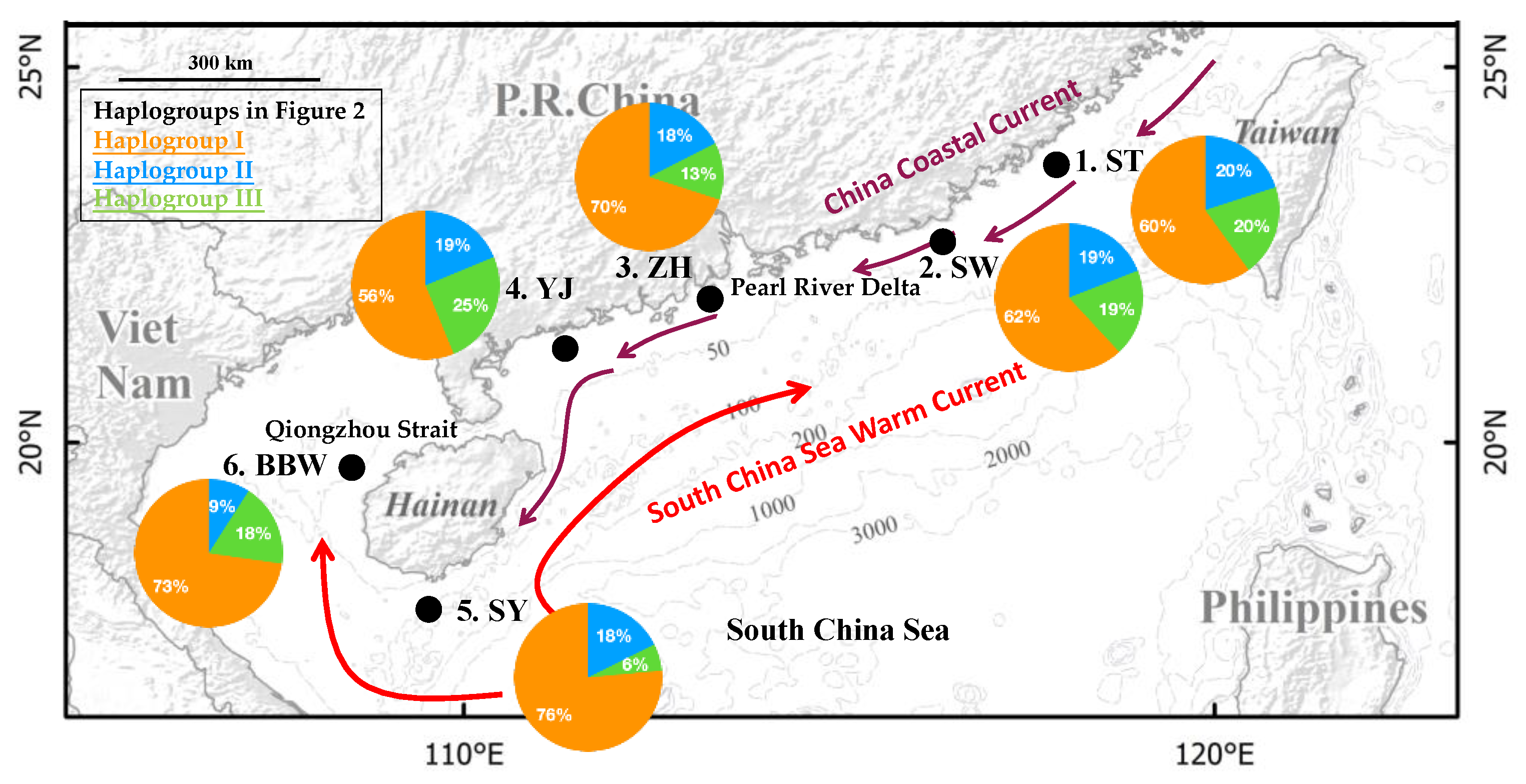
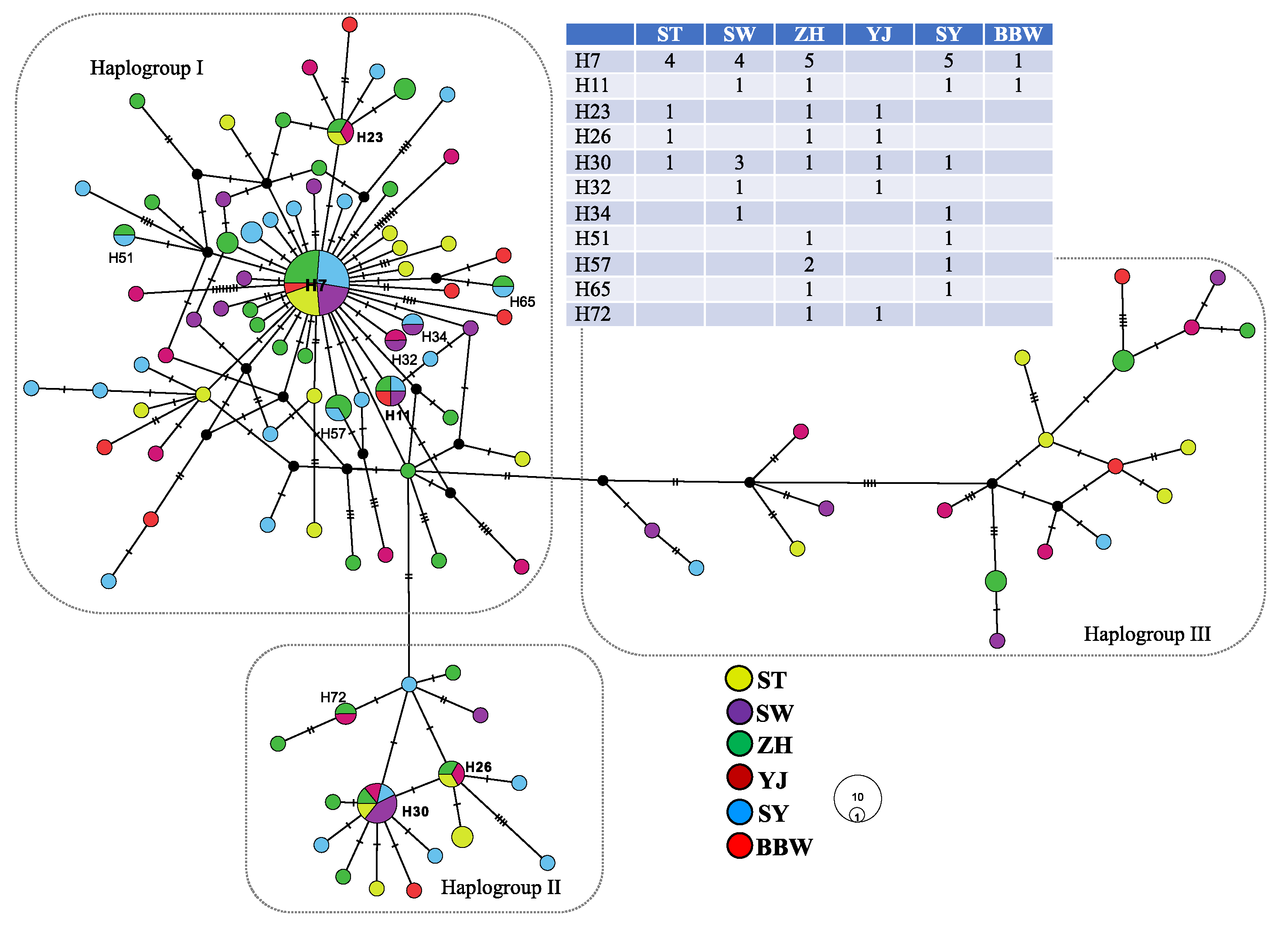
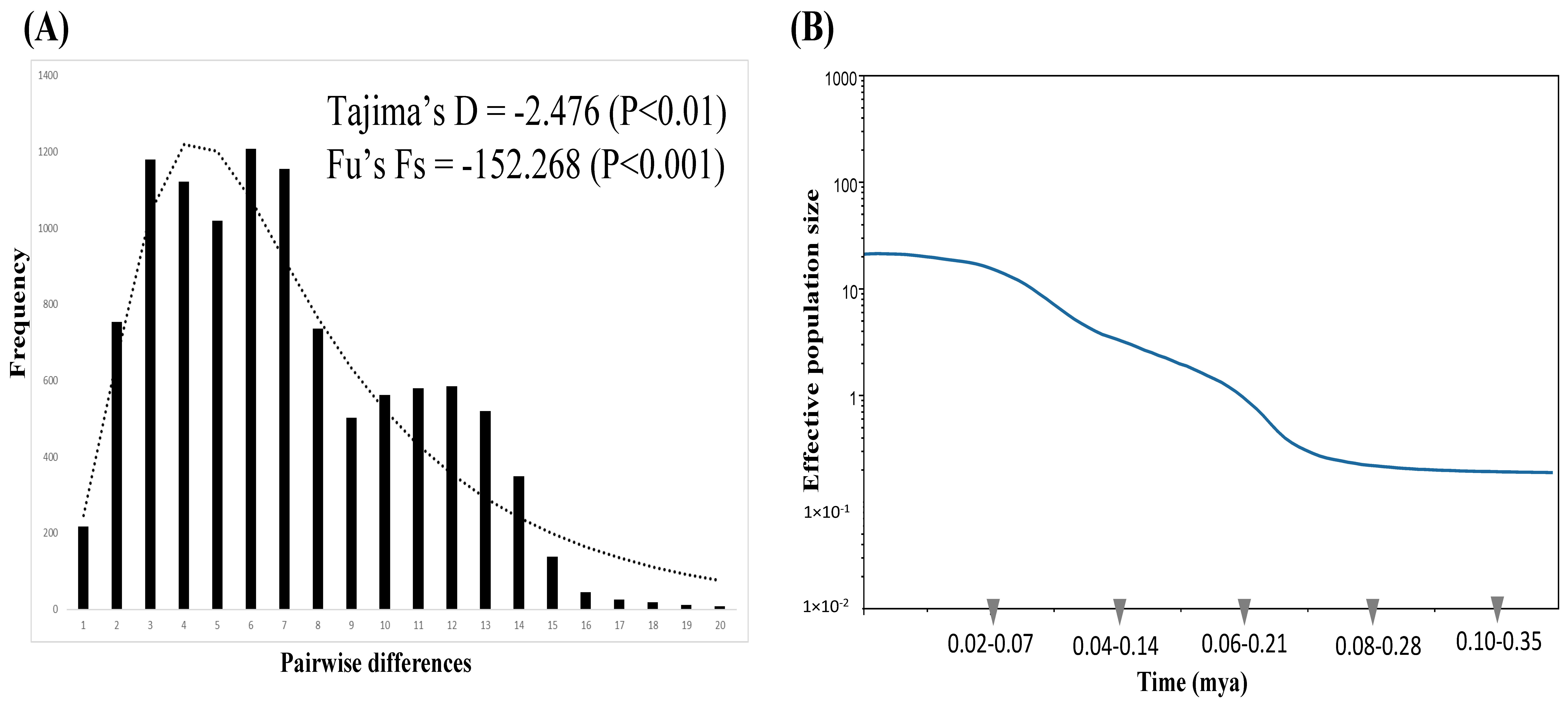

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Code | Date | N | H | Hp | Hs | Hd | π (10−3) |
|---|---|---|---|---|---|---|---|---|
| 1. Shantou | ST | 15 April | 25 | 21 | 17 | 4 | 0.98 | 5.52 |
| 2. Shanwei | SW | 14 April | 21 | 16 | 11 | 5 | 0.96 | 4.62 |
| 3. Zhuhai | ZH | 11 April | 40 | 31 | 22 | 9 | 0.98 | 4.80 |
| 4. Yangjiang | YJ | 11 April | 16 | 16 | 11 | 5 | 1.00 | 8.00 |
| 5. Sanya | SY | 29 March | 34 | 29 | 22 | 7 | 0.98 | 4.36 |
| 6. Beibu Gulf | BBW | 1 January | 11 | 11 | 9 | 2 | 1.00 | 6.69 |
| Total | 147 | 103 | 92 | 11 | 0.98 | 5.26 |
| ST | SW | ZH | YJ | SY | BBW | |
|---|---|---|---|---|---|---|
| ST | 0.500 | 0.512 | 0.663 | 0.502 | 0.599 | |
| SW | 0.000 2 | 0.463 | 0.625 | 0.446 | 0.554 | |
| ZH | 0.000 4 | 0.000 3 | 0.632 | 0.461 | 0.563 | |
| YJ | 0.000 3 | 0.000 2 | 0.000 4 | 0.629 | 0.721 | |
| SY | 0.015 * 2 | 0.000 4 | 0.006 * 6 | 0.077 * 1 | 0.548 | |
| BBW | 0.000 1 | 0.000 2 | 0.000 2 | 0.000 0 | 0.000 2 |
| K | Groupings | FCT | % | D (10−3) |
|---|---|---|---|---|
| 2 | (SY) (BBW,SW,ST,YJ,ZH) | 0.015593 | 1.56 | 0.500 |
| 3 | (SY) (YJ) (BBW,ST,SW,ZH) | 0.016767 | 1.68 | 0.585 |
| 4 | (ST) (SY) (YJ) (BBW,SW,ZH) | 0.0168116 | 1.68 | 0.572 |
| 5 | (ST) (SY) (YJ) (BBW) (SW,ZH) | 0.0194067 | 1.94 | 0.582 |
| 6 | (ST) (SY) (YJ) (BBW) (SW) (ZH) | - | - | 0.561 |
| Scheme | Category Description | % Var. | Statistic | p |
|---|---|---|---|---|
| 1. Three geographic groups: (mainland) (Hainan Island) (Beibu Gulf) | ||||
| Among groups | 1.22 | FSC = 0.00 | <0.578 | |
| Among populations in group | −0.93 | FST = 0.00 | <0.842 | |
| Within population | 99.71 | FCT = 0.01 | <0.136 | |
| 2. Five SAMOVA groups: (ST) (SY) (YJ) (BBW) (SW, ZH) | ||||
| Among groups | 1.94 | FSC = −0.02 | <0.714 | |
| Among populations in groups | −1.98 | FST = 0.00 | <0.572 | |
| Within populations | 100.04 | FCT = 0.02 | <0.065 | |
| 3. Three NETWORK groups in Figure 2 | ||||
| Among groups | 55.37 | FSC = 0.02 | <0.266 | |
| Among populations in group | 1.04 | FST = 0.56 | <0.001 | |
| Within populations | 43.59 | FCT = 0.55 | <0.001 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, M.-R.; Hsu, K.-C.; Wang, J.-X.; Feng, B.; Lin, H.-D.; Yan, Y.-R. Genetic Structure and Diversity of the Yellowbelly Threadfin Bream Nemipterus bathybius in the Northern South China Sea. Diversity 2021, 13, 324. https://doi.org/10.3390/d13070324
Yi M-R, Hsu K-C, Wang J-X, Feng B, Lin H-D, Yan Y-R. Genetic Structure and Diversity of the Yellowbelly Threadfin Bream Nemipterus bathybius in the Northern South China Sea. Diversity. 2021; 13(7):324. https://doi.org/10.3390/d13070324
Chicago/Turabian StyleYi, Mu-Rong, Kui-Ching Hsu, Jin-Xi Wang, Bo Feng, Hung-Du Lin, and Yun-Rong Yan. 2021. "Genetic Structure and Diversity of the Yellowbelly Threadfin Bream Nemipterus bathybius in the Northern South China Sea" Diversity 13, no. 7: 324. https://doi.org/10.3390/d13070324
APA StyleYi, M.-R., Hsu, K.-C., Wang, J.-X., Feng, B., Lin, H.-D., & Yan, Y.-R. (2021). Genetic Structure and Diversity of the Yellowbelly Threadfin Bream Nemipterus bathybius in the Northern South China Sea. Diversity, 13(7), 324. https://doi.org/10.3390/d13070324