DNA Metabarcoding of Deep-Sea Sediment Communities Using COI: Community Assessment, Spatio-Temporal Patterns and Comparison with 18S rDNA

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Area and Sampling
2.2. DNA Extraction, Amplification and Sequencing
2.3. Metabarcoding Pipeline and Taxonomic Assignment
2.4. Data Analysis
2.5. COI-18S Comparison
3. Results
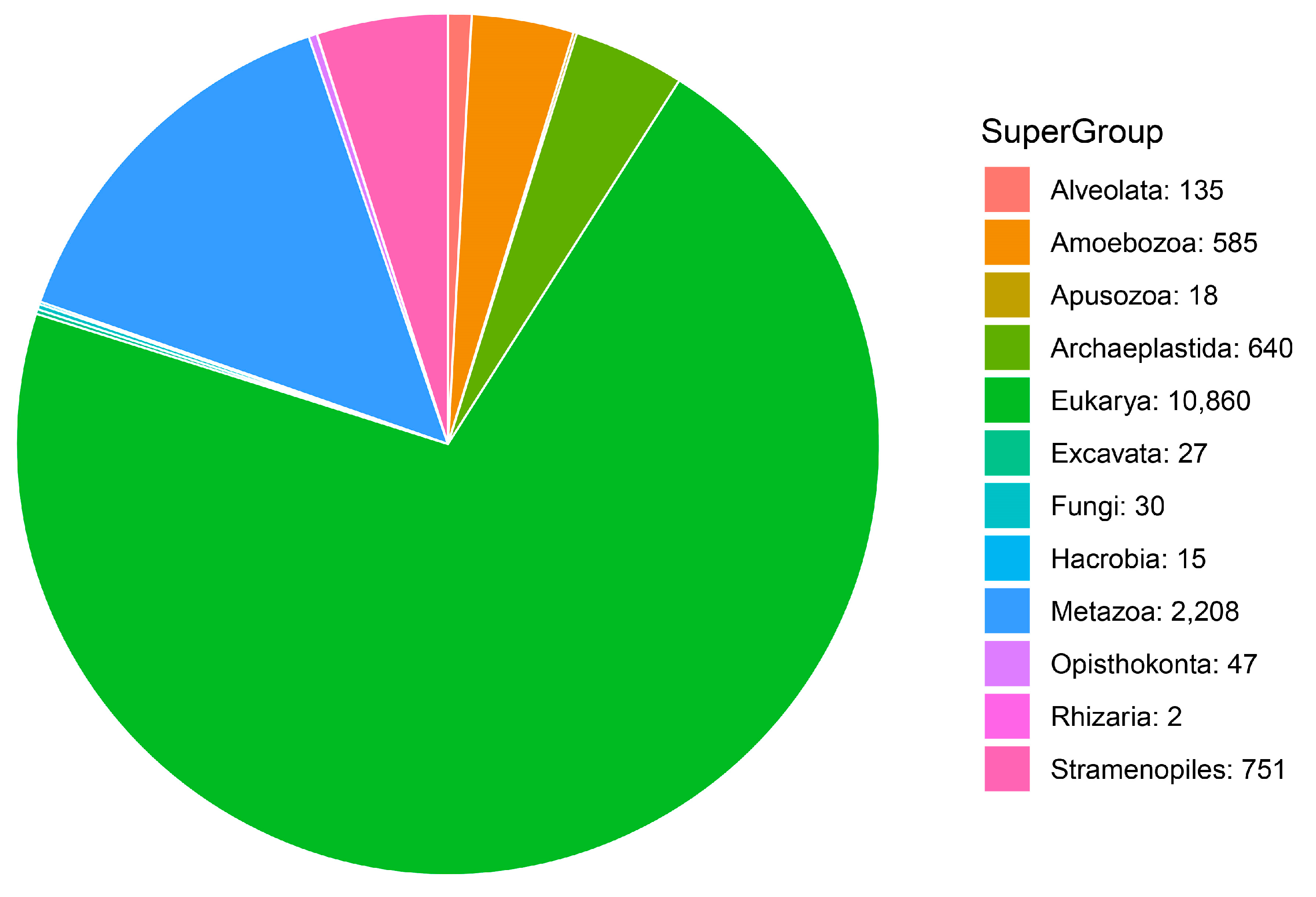
3.1. Community Profile
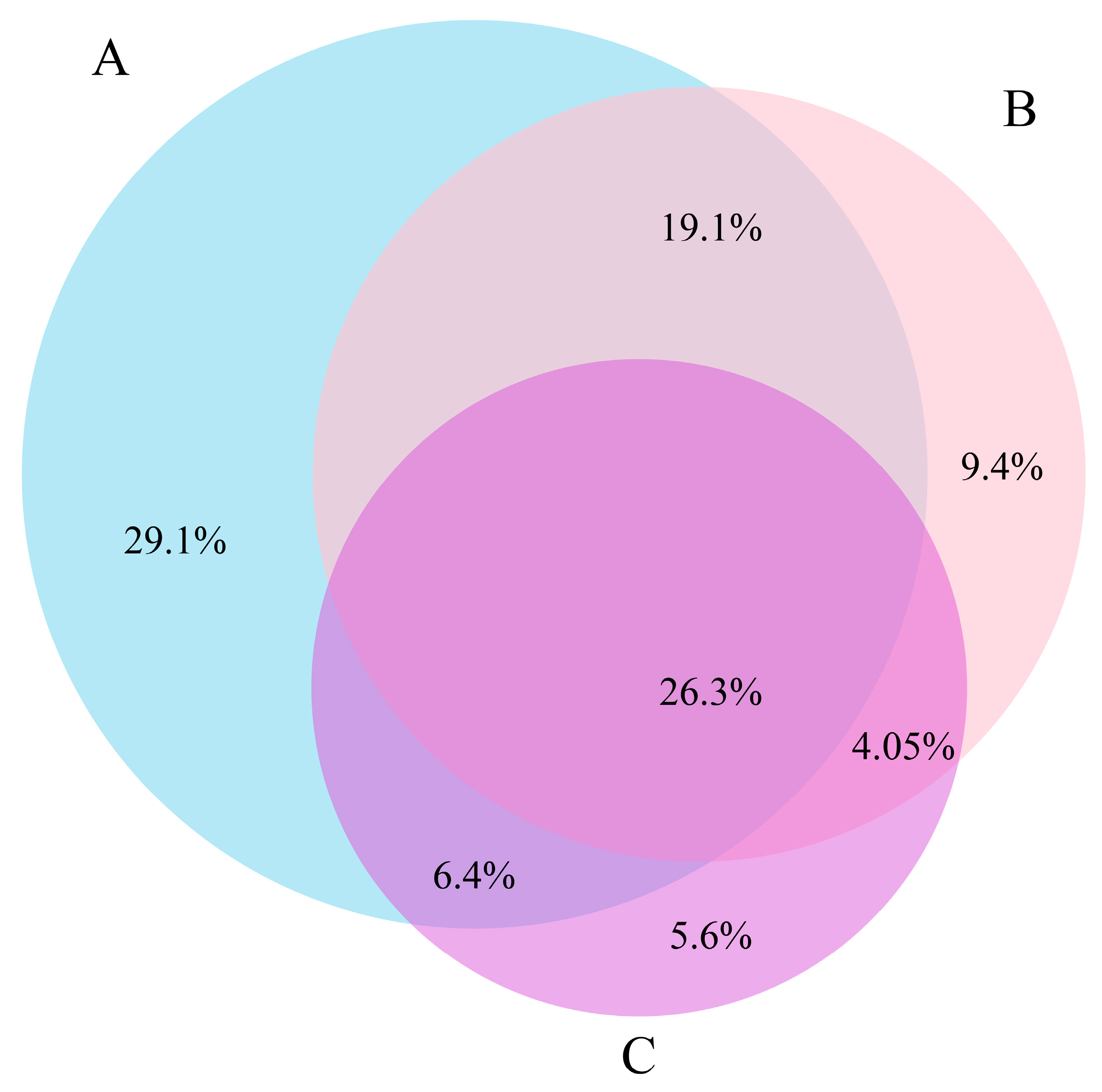
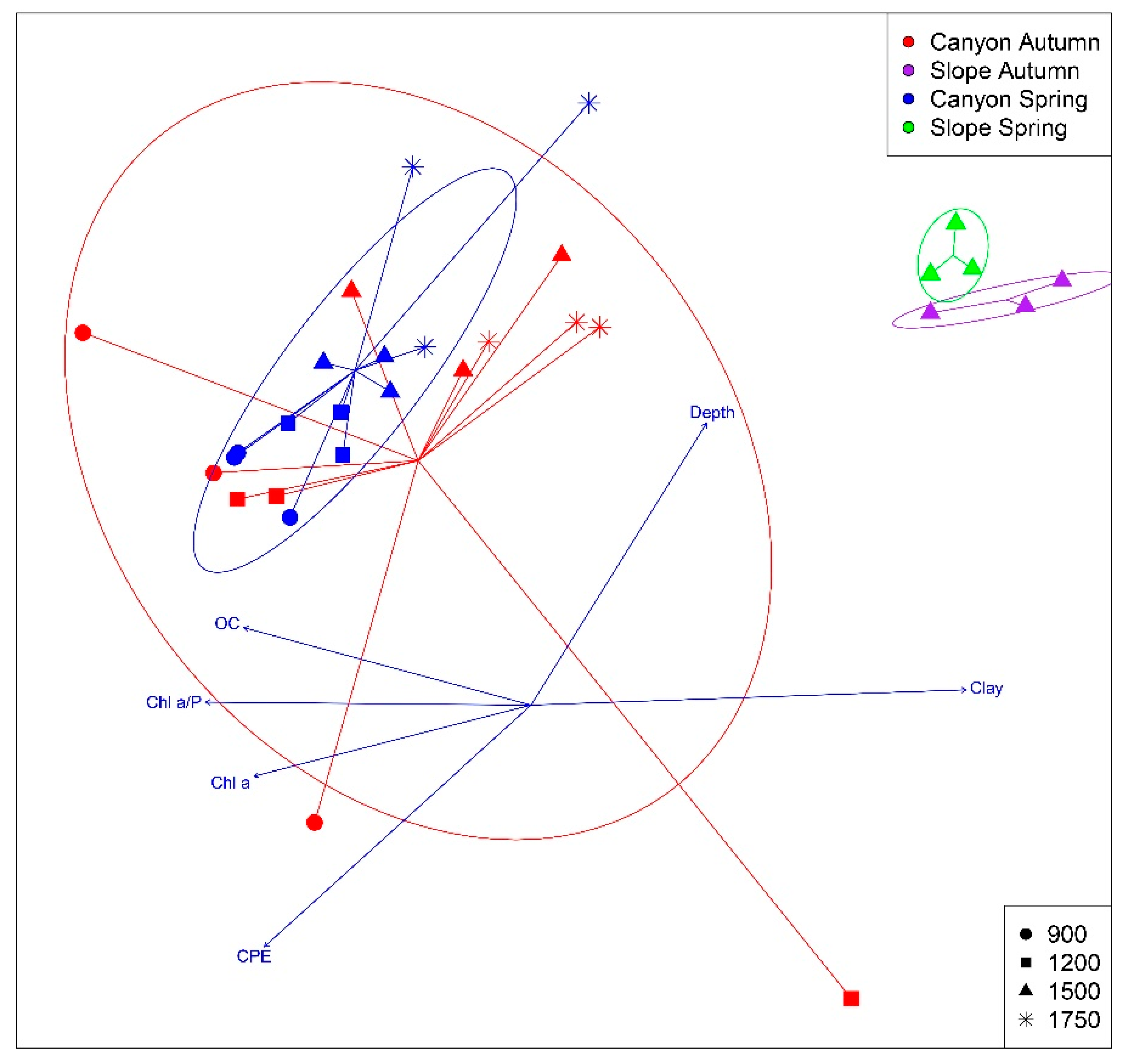
3.2. Spatio-temporal Patterns
3.3. COI-18S Comparison
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eakins, B.W.; Sharman, G.F. Volumes of the World’s Oceans from ETOPO1. NOAA National Geophysical Data Center, Boulder, CO. 2010. Available online: https://www.ngdc.noaa.gov/mgg/global/etopo1_ocean_volumes.html (accessed on 16 November 2019).
- Costello, M.J.; Cheung, A.; De Hauwere, N. Topography statistics for the surface and seabed area, volume, depth and slope, of the world’s seas, oceans and countries. Environ. Sci. Technol. 2010, 44, 8821–8828. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Llodra, E.; Tyler, P.A.; Baker, M.C.; Bergstad, O.A.; Clark, M.R.; Escobar, E.; Levin, L.A.; Lenaick, M.; Ashley, A.R.; Craig, R.S.; et al. Man and the last great wilderness: Human impact on the deep sea. PLoS ONE 2011, 6, e22588. [Google Scholar] [CrossRef] [PubMed]
- Danovaro, R.; Snelgrove, P.V.R.; Tyler, P. Challenging the paradigms of deep-sea ecology. Trends Ecol. Evol. 2014, 29, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Danovaro, R.; Fanelli, E.; Canals, M.; Ciuffardi, T.; Fabri, M.-C.; Taviani, M.; Argyrou, M.; Azzurro, E.; Bianchelli, S.; Cantafaro, A.; et al. Towards a marine strategy for the deep Mediterranean Sea: Analysis of current ecological status. Mar. Policy 2020, 112. [Google Scholar] [CrossRef]
- Coll, M.; Piroddi, C.; Steenbeek, J.; Kaschner, K.; Ben Rais Lasram, F.; Aguzzi, J.; Ballesteros, E.; Bianchi, C.N.; Corbera, J.; Dailianis, T.; et al. The Biodiversity of the Mediterranean Sea: Estimates, Patterns, and Threats. PLoS ONE 2010, 5, e11842. [Google Scholar] [CrossRef]
- Danovaro, R.; Company, J.B.; Corinaldesi, C.; D’Onghia, G.; Galil, B.; Gambi, C.; Gooday, A.J.; Lampadariou, N.; Luna, G.M.; Morigi, C.; et al. Deep-Sea Biodiversity in the Mediterranean Sea: The Known, the Unknown, and the Unknowable. PLoS ONE 2010, 5, e11832. [Google Scholar] [CrossRef]
- Harris, P.; Whiteway, T. Global distribution of large submarine canyons: Geomorphic differences between active and passive continental margins. Mar. Geol. 2011, 285, 69–86. [Google Scholar] [CrossRef]
- Guillén, J.; Bourrin, F.; Palanques, A.; Durrieu de Madron, X.; Puig, P.; Buscail, R. Sediment dynamics during wet and dry storm events on the Têt inner shelf (SW Gulf of Lions). Mar. Geol. 2006, 234, 129–142. [Google Scholar] [CrossRef]
- Bonnin, J.; Heussner, S.; Calafat, A.; Fabres, J.; Palanques, A.; Durrieu de Madron, X.; Canals, M.; Puig, P.; Avril, J.; Delsaut, N. Comparison of horizontal and downward particle fluxes across canyons of the Gulf of Lions (NW Mediterranean): Meteorological and hydrodynamical forcing. Cont. Shelf Res. 2008, 28, 1957–1970. [Google Scholar] [CrossRef]
- Canals, M.; Puig, P.; Durrieu de Madron, X.; Heussner, S.; Palanques, A.; Fabres, J. Flushing submarine canyons. Nature 2006, 444, 354–357. [Google Scholar] [CrossRef]
- Heussner, S.; Durrieu de Madron, X.; Calafat, A.; Canals, M.; Carbonne, J.; Delsaut, N.; Saragoni, G. Spatial and temporal variability of downward particle fluxes on a continental slope: Lessons from an 8-yr experiment in the Gulf of Lions (NW Mediterranean). Mar. Geol. 2006, 234, 63–92. [Google Scholar] [CrossRef]
- Ulses, C.; Estournel, C.; Puig, P.; Durrieu de Madron, X.; Marsaleix, P. Dense shelf water cascading in the northwestern Mediterranean during the cold winter 2005: Quantification of the export through the Gulf of Lion and the Catalan margin. Geophys. Res. Lett. 2008, 35. [Google Scholar] [CrossRef]
- Canals, M.; Company, J.B.; Martín, D.; Sànchez-Vidal, A.; Ramírez-Llodra, E. Integrated study of Mediterranean deep canyons: Novel results and future challenges. Prog. Oceanogr. 2013, 118. [Google Scholar] [CrossRef]
- Lopez-Fernandez, P.; Calafat, A.; Sanchez-Vidal, A.; Canals, M.; Mar Flexas, M.; Cateura, J.; Company, J.B. Multiple drivers of particle fluxes in the Blanes submarine canyon and southern open slope: Results of a year round experiment. Prog. Oceanogr. 2013, 118, 95–107. [Google Scholar] [CrossRef]
- Puig, P.; Martín, J.; Masqué, P.; Palanques, A. Increasing sediment accumulation rates in La Fonera (Palamós) submarine canyon axis and their relationship with bottom trawling activities. Geophys. Res. Lett. 2015, 42, 8106–8113. [Google Scholar] [CrossRef]
- Paradis, S.; Puig, P.; Sanchez-Vidal, A.; Masqué, P.; Garcia-Orellana, J.; Calafat, A.; Canals, M. Spatial distribution of sedimentation-rate increases in Blanes Canyon caused by technification of bottom trawling fleet. Prog. Oceanogr. 2018, 169, 241–252. [Google Scholar] [CrossRef]
- Zúñiga, D.; Flexas, M.M.; Sanchez-Vidal, A.; Coenjaerts, J.; Calafat, A.; Jordà, G.; Company, J.B. Particle fluxes dynamics in Blanes submarine canyon (Northwestern Mediterranean). Prog. Oceanogr. 2009, 82, 239–251. [Google Scholar] [CrossRef]
- Snelgrove, P.V.R. An ocean of discovery: Biodiversity beyond the census of marine life. Planta Med. 2016, 82, 790–799. [Google Scholar] [CrossRef]
- Bohmann, K.; Evans, A.; Gilbert, M.T.P.; Carvalho, G.R.; Creer, S.; Knapp, M.; Yu, D.W.; de Bruyn, M. Environmental DNA for wildlife biology and biodiversity monitoring. Trends Ecol. Evol. 2014, 29, 358–367. [Google Scholar] [CrossRef]
- Creer, S.; Deiner, K.; Frey, S.; Porazinska, D.; Taberlet, P.; Thomas, W.K.; Potter, C.; Bik, H.M. The ecologist’s field guide to sequence-based identification of biodiversity. Methods Ecol. Evol. 2016, 7, 1008–1018. [Google Scholar] [CrossRef]
- Lodge, D.M.; de Vere, N.; Pfrender, M.E.; Bematchez, L. Environmental DNA metabarcoding: Transforming how we survey animal and plant communities. Mol. Ecol. 2017, 26, 5872–5895. [Google Scholar] [CrossRef]
- Sinniger, F.; Pawlowski, J.; Harii, S.; Gooday, A.J.; Yamamoto, H.; Chevaldonné, P.; Cedhagen, T.; Carvalho, G.; Creer, S. Worldwide Analysis of Sedimentary DNA Reveals Major Gaps inTaxonomic Knowledge of Deep-SeaBenthos. Front. Mar. Sci. 2016, 3, 92. [Google Scholar] [CrossRef]
- Brandt, M.I.; Trouche, B.; Quintric, L.; Wincker, P.; Poulain, J.; Arnaud-Haond, S. A flexible pipeline combining bioinformatic correction tools for prokaryotic and eukaryotic metabarcoding. bioRxiv 2019. [Google Scholar] [CrossRef]
- Taberlet, P.; Coissac, E.; Pompanon, F.; Brochmann, C.; Willerslev, E. Towards next-generation biodiversity assessment using DNA metabarcoding. Mol. Ecol. 2012, 21, 2045–2050. [Google Scholar] [CrossRef] [PubMed]
- Dell’Anno, A.; Danovaro, R. Extracellular DNA plays a key role in deep-sea ecosystem functioning. Science 2005, 309, 2179. [Google Scholar] [CrossRef] [PubMed]
- Stoeck, T.; Behnke, A.; Christen, R.; Amaral-Settler, L.; Rodriguez-Mora, M.J.; Chistoserdov, A.; Orsi, W.; Edgcomb, V.P. Massively parallel tag sequencing reveals the complexity of anaerobic marine protistan communities. BMC Biol. 2009, 7, 72. [Google Scholar] [CrossRef]
- Pawlowski, J.; Christen, R.; Lecroq, B.; Bachar, D.; Shahbazkia, H.R.; Amaral-Zettler, L.; Guillou, L. Eukaryotic Richness in the Abyss: Insights from Pyrotag Sequencing. PLoS ONE 2011, 6, e18169. [Google Scholar] [CrossRef]
- Bik, H.M.; Porazinska, D.L.; Creer, S.; Caporaso, J.G.; Knight, R.; Thomas, W.K. Sequencing our way towards understanding global eukaryotic biodiversity. Trends Ecol. Evol. 2012, 27, 233–243. [Google Scholar] [CrossRef]
- Guardiola, M.; Uriz, M.J.; Taberlet, P.; Coissac, E.; Wangensteen, O.S.; Turon, X. Deep-Sea, Deep-Sequencing: Metabarcoding Extracellular DNA from Sediments of Marine Canyons. PLoS ONE 2015, 10, e0139633. [Google Scholar] [CrossRef]
- Guardiola, M.; Wangensteen, O.S.; Taberlet, P.; Coissac, E.; Uriz, M.J.; Turon, X. Spatio-temporal monitoring of deep-sea communities using metabarcoding of sediment DNA and RNA. PeerJ 2016, 4, e2807. [Google Scholar] [CrossRef]
- Brannock, P.M.; Learman, D.R.; Mahon, A.R.; Santos, S.R.; Halanych, K.M. Meiobenthic community composition and biodiversity along a 5500 km transect of Western Antarctica: A metabarcoding analysis. Mar. Ecol. Prog. Ser. 2018, 603, 47–60. [Google Scholar] [CrossRef]
- Wangensteen, O.S.; Palacín, C.; Guardiola, M.; Turon, X. DNA metabarcoding of littoral hard-bottom communities: High diversity and database gaps revealed by two molecular markers. PeerJ 2018, 6, e4705. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Chilton, N.B.; Zhu, X.; Gasser, R.B. Single-strand conformation polymorphism-based analysis of mitochondrial cytochrome c oxidase subunit 1 reveals significant substructuring in hookworm populations. Electrophoresis 2002, 23, 27–34. [Google Scholar] [CrossRef]
- Tang, C.Q.; Leasi, F.; Obertegger, U.; Kieneke, A.; Barraclough, T.G.; Fontaneto, D. The widely used small subunit 18S rDNA molecule greatly underestimates true diversity in biodiversity surveys of the meiofauna. Proc. Natl. Acad. Sci. USA 2012, 109, 16208–16212. [Google Scholar] [CrossRef] [PubMed]
- Cowart, D.A.; Pinheiro, M.; Mouchel, O.; Maguer, M.; Grall, J.; Miné, J.; Arnaud-Haond, S. Metabarcoding is powerful yet still blind: A comparative analysis of morphological and molecular surveys of seagrass communities. PLoS ONE 2015, 10, e0117562. [Google Scholar] [CrossRef] [PubMed]
- Turon, X.; Antich, A.; Palacín, C.; Præbel, K.; Wangensteen, O.S. From metabarcoding to metaphylogeography: Separating the wheat from the chaff. Ecol. Appl. 2019, 30, e02036. [Google Scholar] [CrossRef] [PubMed]
- Leray, M.; Yang, J.Y.; Meyer, C.P.; Mills, S.C.; Agudelo, N.; Ranwez, V.; Boehm, J.; Machida, R.J. A new versatil primer set targeting a short fragment of the mithocondrial COI region for metabarcoding metazoan diversity: Application for characterizing coral reef fish gut contents. Front. Zool. 2013, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Ratnasingham, S.; Hebert, P.D.N. Bold: The Barcode of Life Data System (www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef]
- Lastras, G.; Canals, M.; Amblas, D.; Lavoie, C.; Church, I.; De Mol, B.; Duran, R.; Calafat, A.M.; Hughes-Clarke, J.E.; Smith, C.J.; et al. Understanding sediment dynamics of two large submarine valleys from seafloor data: Blanes and La Fonera canyons, northwestern Mediterranean Sea. Mar. Geol. 2011, 280, 20–39. [Google Scholar] [CrossRef]
- Amblas, D.; Canals, M.; Urgeles, R.; Lastras, G.; Liquete, C.; Hughes-Clarke, J.E.; Calafat, A.M. Morphogenetic mesoscale analysis of the northeastern Iberian margin, NW Mediterranean Basin. Mar. Geol. 2006, 234, 3–20. [Google Scholar] [CrossRef]
- Geller, J.; Meyer, C.; Parker, M.; Hawk, H. Redesign of PCR primers for mitochondrial cytochrome c oxidase subunit I for marine invertebrates and application in all-taxa biotic surveys. Mol. Ecol. Resour. 2013, 13, 851–886. [Google Scholar] [CrossRef] [PubMed]
- Boyer, F.; Mercier, C.; Bonin, A.; Le Bras, Y.; Taberlet, P.; Coissac, E. obitools: A unix-inspired software package for DNA metabarcoding. Mol. Ecol. Resour. 2016, 16, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Mendeley Dataset. Available online: https://data.mendeley.com/datasets/7hh8gykjg5/1 (accessed on 28 January 2020).
- Mahé, F.; Rognes, T.; Quince, C.; de Vargas, C.; Dunthorn, M. Swarm v2: Highly-scalable and high-resolution amplicon clustering. PeerJ 2015, 3, e1420. [Google Scholar] [CrossRef] [PubMed]
- Wangensteen, O.S.; Turon, X. Metabarcoding Techniques for Assessing Biodiversity of Marine Animal Forests. In Marine Animal Forests; Rossi, S., Bramanti, L., Gori, A., Orejas, C., Eds.; Springer: Cham, Switzerland, 2017; pp. 445–473. [Google Scholar] [CrossRef]
- Github Repository. Available online: https://github.com/metabarpark/Reference-databases (accessed on 16 November 2019).
- Guillou, L.; Bachar, D.; Audic, S.; Bass, D.; Berney, C.; Bittner, L.; Boutte, C.; Burgaud, G.; De Vargas, C.; Decelle, J.; et al. The protist ribosomal reference database (PR2): A catalog of unicellular eukaryote small subunit rRNA sequences with curated taxonomy. Nucleic Acids Res. 2013, 41, D597–D604. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community Ecology Package. R-Package Version 2.3-4. 2016. Available online: https://cran.r-project.org/package=vegan (accessed on 20 December 2019).
- Fox, J.; Weisberg, S. An R companion to Applied Regression, 3rd ed.; Sage: Thousand Oakes, CA, USA, 2019. [Google Scholar]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef]
- Venables, W.; Ripley, B. Modern Applied Statistics with S. In Statistics and Computing, 4th ed.; Springer-Verlag: Berlin, Germany, 2002. [Google Scholar] [CrossRef]
- Github Repository. Available online: https://github.com/pmartinezarbizu/pairwiseAdonis (accessed on 20 December 2019).
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar]
- Román, S.; Vanreusel, A.; Romano, C.; Ingels, J.; Puig, P.; Company, J.B.; Martin, D. High spatiotemporal variability in meiofaunal assemblages in Blanes Canyon (NW Mediterranean) subject to anthropogenic and natural disturbances. Deep Sea Res. Part I 2016, 117, 70–83. [Google Scholar] [CrossRef]
- Kudryavtsev, A. Paravannella minima n. g. n. sp. (Discosea, Vannellidae) and distinction of the genera in the vannellid amoebae. Eur. J. Protistol. 2014, 50, 258–269. [Google Scholar] [CrossRef]
- Tekle, Y.I. DNA barcoding in amoebozoa and challenges: The example of Cochliopodium. Protist 2014, 165, 473–484. [Google Scholar] [CrossRef]
- Kudryavtsev, A.; Pawlowski, J. Cunea n. g. (Amoebozoa, Dactylopodida) with two cryptic species isolated from different areas of the ocean. Eur. J. Protistol. 2015, 51, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Garcés-Pastor, S.; Wangensteen, O.S.; Pérez-Haase, A.; Pèlachs, A.; Pérez-Obiol, R.; Cañellas-Boltà, N.; Mariani, S.; Vegas-Vilarrúbia, T. DNA metabarcoding reveals modern and past eukaryotic communities in a high mountain peat bog system. J. Paleolimnol. 2019, 62, 425–441. [Google Scholar] [CrossRef]
- Bakker, J.; Wangensteen, O.S.; Baillie, C.; Buddo, D.; Chapman, D.D.; Gallagher, A.; Guttridge, T.L.; Hertler, H.; Mariani, S. Biodiversity assessment of tropical shelf eukaryotic communities via pelagic eDNA metabarcoding. Ecol. Evol. 2019, 9, 14341–14355. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Llodra, E.; Brandt, A.; Danovaro, R.; De Mol, B.; Escobar, E.; German, C.R.; Levin, L.A.; Martinez Arbizu, P.; Menot, L.; Buhl-Mortensen, P.; et al. Deep, diverse and definitely different: Unique attributes of the world’s largest ecosystem. Biogeosciences 2010, 7, 2851–2899. [Google Scholar] [CrossRef]
- Romano, C.; Coenjaerts, J.; Flexas, M.M.; Zúniga, D.; Vanreusel, A.; Company, J.B.; Martín, D. Spatial and temporal variability of meiobenthic density in the Blanes submarine canyon (NW Mediterranean). Prog. Oceanogr. 2013, 118, 144–158. [Google Scholar] [CrossRef]
- Kalogeropoulou, V.; Bett, B.J.; Gooday, A.J.; Lampadariou, N.; Martinez Arbizu, P.; Vanreuselm, A. Temporal changes (1989–1999) in deep-sea metazoan meiofaunal assemblages on the Porcupine Abyssal Plain, NE Atlantic. Deep Sea Res. Part II 2016, 57, 1383–1395. [Google Scholar] [CrossRef][Green Version]
- Román, S.; Lins, L.; Ingels, J.; Romano, C.; Martin, D.; Vanreusel, A. Role of spatial scales and environmental drivers in shaping nematode communities in the Blanes Canyon and its adjacent slope. Deep Sea Res Part I 2019, 146, 62–78. [Google Scholar] [CrossRef]
- Tecchio, S.; Ramírez-Llodra, E.; Aguzzi, J.; Sanchez-Vidal, A.; Flexas, M.M.; Sardà, F.; Company, J.B. Seasonal fluctuations of deep megabenthos: Finding evidence of standing stock accumulation in a flux-rich continental slope. Prog. Oceanogr. 2013, 118, 188–198. [Google Scholar] [CrossRef]
- Fernandez-Arcaya, U.; Rotllant, G.; Ramirez-Llodra, E.; Recasens, L.; Aguzzi, J.; Flexas, M.M.; Sanchez-Vidal, A.; López-Fernández, P.; García, J.A.; Company, J.B. Reproductive biology and recruitment of the deep-sea fish community from the NW Mediterranean continental margin. Prog. Oceanogr. 2013, 118, 222–234. [Google Scholar] [CrossRef]
- Gooday, A.J. Biological Responses to Seasonally Varying Fluxes of Organic Matter to the Ocean Floor: A Review. J. Oceanogr. 2002, 58, 305–332. [Google Scholar] [CrossRef]
- Ramirez-Llodra, E.; Company, J.B.; Sardà, F.; Rotllant, G. Megabenthic diversity patterns and community structure of the Blanes submarine canyon and adjacent slope in the Northwestern Mediterranean: A human overprint? Mar. Ecol. 2010, 31, 167–182. [Google Scholar] [CrossRef]
- Costello, M.J.; Chaudhary, C. Marine Biodiversity, Biogeography, Deep-Sea Gradients, and Conservation. Curr. Biol. 2017, 27, R511–R527. [Google Scholar] [CrossRef] [PubMed]
- Danovaro, R.; Tselepides, A.; Otegui, A.; Della Croce, N. Dynamics of meiofaunal assemblages on the continental shelf and deep-sea sediments of the Cretan Sea (NE Mediterranean): Relationships with seasonal changes in food supply. Prog. Oceanogr. 2000, 46, 367–400. [Google Scholar] [CrossRef]
- Etter, R.J.; Grassle, J.F. Patterns of species diversity in the deep sea as a function of sediment particle size diversity. Nature 1992, 360, 576–578. [Google Scholar] [CrossRef]
- Leduc, D.; Rowden, A.A.; Probert, P.K.; Pilditch, C.A.; Nodder, S.D.; Vanreusel, A.; Duineveld, G.C.A.; Witbaard, R. Further evidence for the effect of particle-size diversity on deep-sea benthic biodiversity. Deep Sea Res. Part I 2012, 63, 164–169. [Google Scholar] [CrossRef]
- Stefanni, S.; Stanković, D.; Borme, D.; de Olazabal, A.; Juretic, T.; Pallavicini, A.; Tirelli, V. Multi-marker metabarcoding approach to study mesozooplankton at basin scale. Sci. Rep. 2018, 8, 12085. [Google Scholar] [CrossRef]
- Leray, M.; Knowlton, N. Censusing marine eukaryotic diversity in the twenty-first century. Philos. Trans. R. Soc. B 2016, 371, 20150331. [Google Scholar] [CrossRef]
- Hadziavdic, K.; Lekang, K.; Lanzen, A.; Jonassen, I.; Thompson, E.M.; Troedsson, C. Characterization of the 18S rRNA Gene for Designing Universal Eukaryote Specific Primers. PLoS ONE 2014, 9, e87624. [Google Scholar] [CrossRef]
- Morard, R.; Lejzerowicz, F.; Darling, K.; Lecroq-Bennet, B.; Pedersen, M.; Orlando, L.; Pawlowski, J.; Mulitza, S.; de Vargas, C.; Kucera, M. Plankton-derived environmental DNA extracted from abyssal sediments preserves patterns of plankton macroecology. Biogeosciences 2017, 14, 2741–2754. [Google Scholar] [CrossRef]
- Corinaldesi, C.; Barucca, M.; Luna, G.M.; Dell’Anno, A. Preservation, origin and genetic imprint of extracellular DNA in permanently anoxic deep-sea sediments. Mol. Ecol. 2011, 20, 642–654. [Google Scholar] [CrossRef]
- Lejzerowicz, F.; Esling, P.; Majewski, W.; Szczucinski, W.; Decelle, J.; Obadia, C.; Arbizu, P.M.; Pawlowski, J. Ancient DNA complements microfossil record in deep-sea subsurface sediments. Biol. Lett. 2013, 9, 20130283. [Google Scholar] [CrossRef] [PubMed]
- Torti, A.; Lever, M.A.; Jørgensen, B.B. Origin, dynamics, and implications of extracellular DNA pools in marine sediments. Mar. Genom. 2015, 24, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Deagle, B.E.; Jarman, S.N.; Coissac, E.; Pompanon, F.; Taberlet, P. DNA metabarcoding and the cytochrome c oxidase subunit I marker: Not a perfect match. Biol. Lett. 2014, 10. [Google Scholar] [CrossRef]
- Elbrecht, V.; Leese, F. Validation and development of COI metabarcoding primers for freshwater macroinvertebrate bioassessment. Front. Environ. Sci. 2017, 5, 11. [Google Scholar] [CrossRef]
- Vamos, E.E.; Elbrecht, V.; Leese, F. Short COI markers for freshwater macroinvertebrate metabarcoding. MBMG 2017, 1, e14625. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DOSMARES II CRUISE | |||||||
|---|---|---|---|---|---|---|---|
| Zone | Date | Station | Depth (m) | Lat N | Long E | ||
| Blanes Canyon (BC) | 07/10/2012 | BC900 | 874 | 41°31′17″ | 02°50′52′ | ||
| 890 | 41°34′14″ | 02°50′47″ | |||||
| 852 | 41°34′17″ | 02°50′35″ | |||||
| BC1200 | 1232 | 41°30′43″ | 02°50′46″ | ||||
| 1194 | 41°30′44″ | 02°50′35″ | |||||
| 08/10/2012 | 1248 | 41°30′44″ | 02°50′54″ | ||||
| BC1500 | 1450 | 41°27′31″ | 02°52′57″ | ||||
| 1463 | 41°27′38″ | 02°52′46″ | |||||
| 1457 | 41°27′29″ | 02°52′58″ | |||||
| 11/10/2012 | BC1750 | 1746 | 41°21′16″ | 02°52′13″ | |||
| 1751 | 41°21′20″ | 02°52′13″ | |||||
| 1727 | 41°21′38″ | 02°52′15″ | |||||
| Open Slope (OS) | 12/10/2012 | OS1500 | 1454 | 41°08′42″ | 02°53′33″ | ||
| 1451 | 41°08′37″ | 02°53′32″ | |||||
| 1480 | 41°08′30.7″ | 02°53′48.1″ | |||||
| DOSMARES III CRUISE | |||||||
| Zone | Date | Station | Depth (m) | Lat N | Long E | ||
| Blanes Canyon (BC) | 5/4/2013 | BC900 | 870 | 41°34′19″ | 02°50′59″ | ||
| 903 | 41°34′19″ | 02°50′56″ | |||||
| 899 | 41°34′19″ | 02°50′53″ | |||||
| 6/4/2013 | BC1200 | 1227 | 41°31′01″ | 02°51′03″ | |||
| 1258 | 41°30′52″ | 02°50′50″ | |||||
| 05/04/2013 | 1234 | 41°30′46″ | 02°50′54″ | ||||
| BC1500 | 1483 | 42°27′15″ | 02°52′57″ | ||||
| 23/04/2013 | 1516 | 41°27′03″ | 02°52′53″ | ||||
| 24/04/2013 | 1520 | 41°27′02″ | 02°52′50″ | ||||
| 23/04/2013 | BC1750 | 1730 | 41°21′14″ | 02°51′54″ | |||
| 1785 | 41°21′14″ | 02°52′17″ | |||||
| 1830 | 41°21′06″ | 02°52′20″ | |||||
| Open Slope (OS) | 22/04/2013 | OS1500 | 1507 | 41°08′28″ | 02°53′46″ | ||
| 23/04/2013 | 1480 | 41°08′23″ | 02°53′33″ | ||||
| 1456 | 41°08′18″ | 02°53′41″ | |||||
| Taxonomic Category | Proportion of MOTUs Assigned to Each Category | Super-Group | Proportion of MOTUs Assigned to Species Category |
|---|---|---|---|
| Super-Group | 43.00% | Amoebozoa | 52.32% |
| Phylum | 10.3% | Apusozoa | 2.14% |
| Class | 17.09% | Archaeplastida | 14.51% |
| Order | 8.37% | Alveolata | 3.92% |
| Family | 0.79% | Hacrobia | 0.59% |
| Genus | 1.59% | Rhizaria | 0.24% |
| Species | 18.86% | Stramenopiles | 10.23% |
| Excavata | 0.24% | ||
| Metazoa | 15.58% | ||
| Fungi | 0.24% |
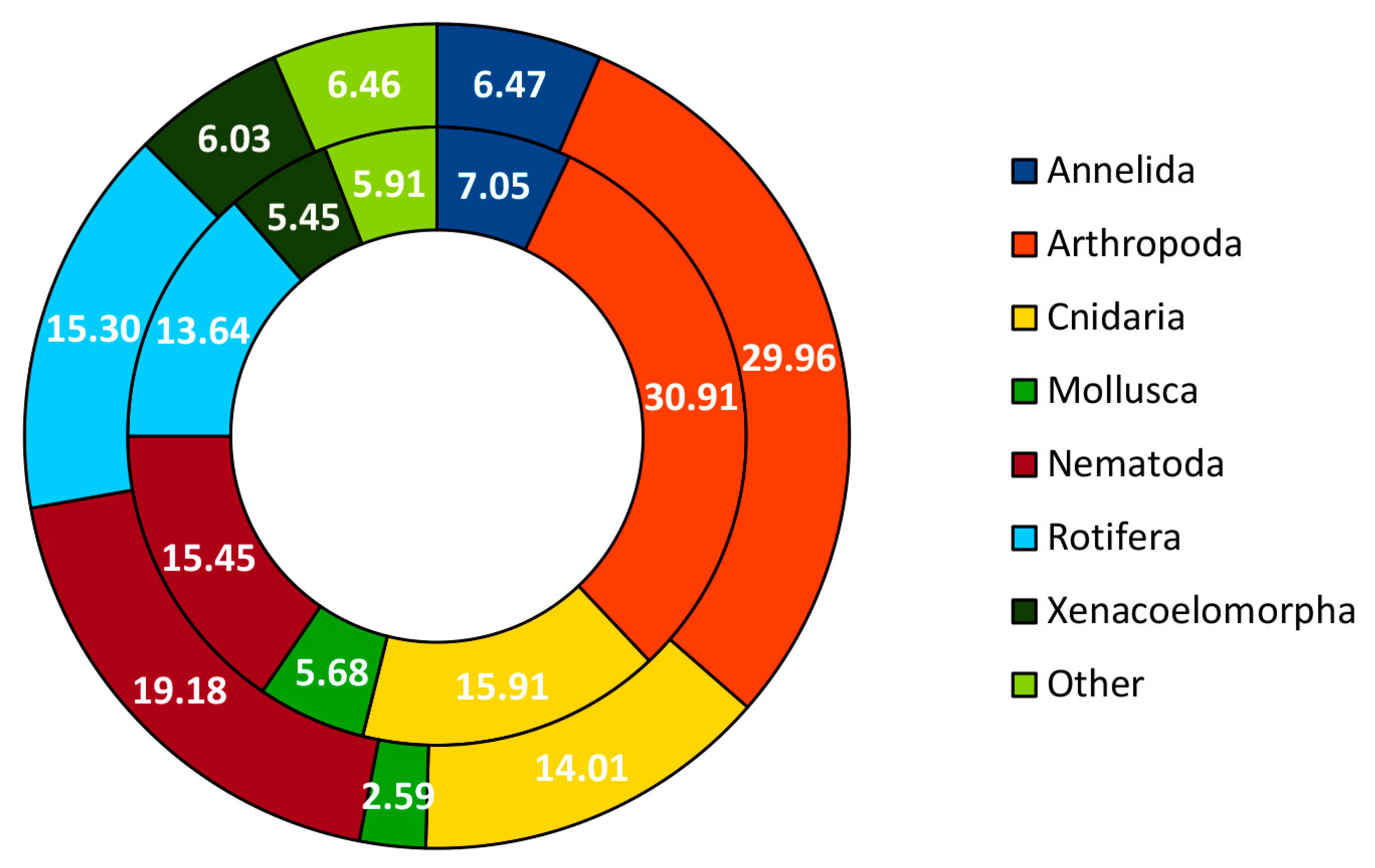
| Taxonomic Category | Proportion of MOTUs Assigned to Each Category | Metazoan Phylum | Proportion of MOTUs Assigned to Species Category |
|---|---|---|---|
| Phylum | 8.36% | Annelida | 2.29% |
| Class | 13.34% | Arthropoda | 7.63% |
| Order | 53.86% | Bryozoa | 1.53% |
| Family | 0.64% | Chordata | 3.05% |
| Genus | 2.73% | Cnidaria | 9.16% |
| Species | 21.06% | Mollusca | 4.58% |
| Nematoda | 0.76% | ||
| Porifera | 4.58% | ||
| Rotifera | 61.07% | ||
| Xenacoelomorpha | 5.34% |
| Variable | r2 | p |
|---|---|---|
| Depth | 0.502 | 0.001 * |
| Clay | 0.681 | 0.001 * |
| CPE | 0.542 | 0.001 * |
| Chl a | 0.301 | 0.015 * |
| Chl a / P | 0.381 | 0.002 * |
| OC | 0.326 | 0.004 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atienza, S.; Guardiola, M.; Præbel, K.; Antich, A.; Turon, X.; Wangensteen, O.S. DNA Metabarcoding of Deep-Sea Sediment Communities Using COI: Community Assessment, Spatio-Temporal Patterns and Comparison with 18S rDNA. Diversity 2020, 12, 123. https://doi.org/10.3390/d12040123
Atienza S, Guardiola M, Præbel K, Antich A, Turon X, Wangensteen OS. DNA Metabarcoding of Deep-Sea Sediment Communities Using COI: Community Assessment, Spatio-Temporal Patterns and Comparison with 18S rDNA. Diversity. 2020; 12(4):123. https://doi.org/10.3390/d12040123
Chicago/Turabian StyleAtienza, Sara, Magdalena Guardiola, Kim Præbel, Adrià Antich, Xavier Turon, and Owen Simon Wangensteen. 2020. "DNA Metabarcoding of Deep-Sea Sediment Communities Using COI: Community Assessment, Spatio-Temporal Patterns and Comparison with 18S rDNA" Diversity 12, no. 4: 123. https://doi.org/10.3390/d12040123
APA StyleAtienza, S., Guardiola, M., Præbel, K., Antich, A., Turon, X., & Wangensteen, O. S. (2020). DNA Metabarcoding of Deep-Sea Sediment Communities Using COI: Community Assessment, Spatio-Temporal Patterns and Comparison with 18S rDNA. Diversity, 12(4), 123. https://doi.org/10.3390/d12040123