Definition of Core Bacterial Taxa in Different Root Compartments of Dactylis glomerata, Grown in Soil under Different Levels of Land Use Intensity

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling Sites
2.2. Sampling and Basic Analyses
2.3. Nucleic Acid Extraction
2.4. Library Preparation and Illumina Sequencing
2.5. Sequence Data Analysis
3. Results
3.1. Soil Carbon and Nitrogen Content
3.2. Sequencing Summary
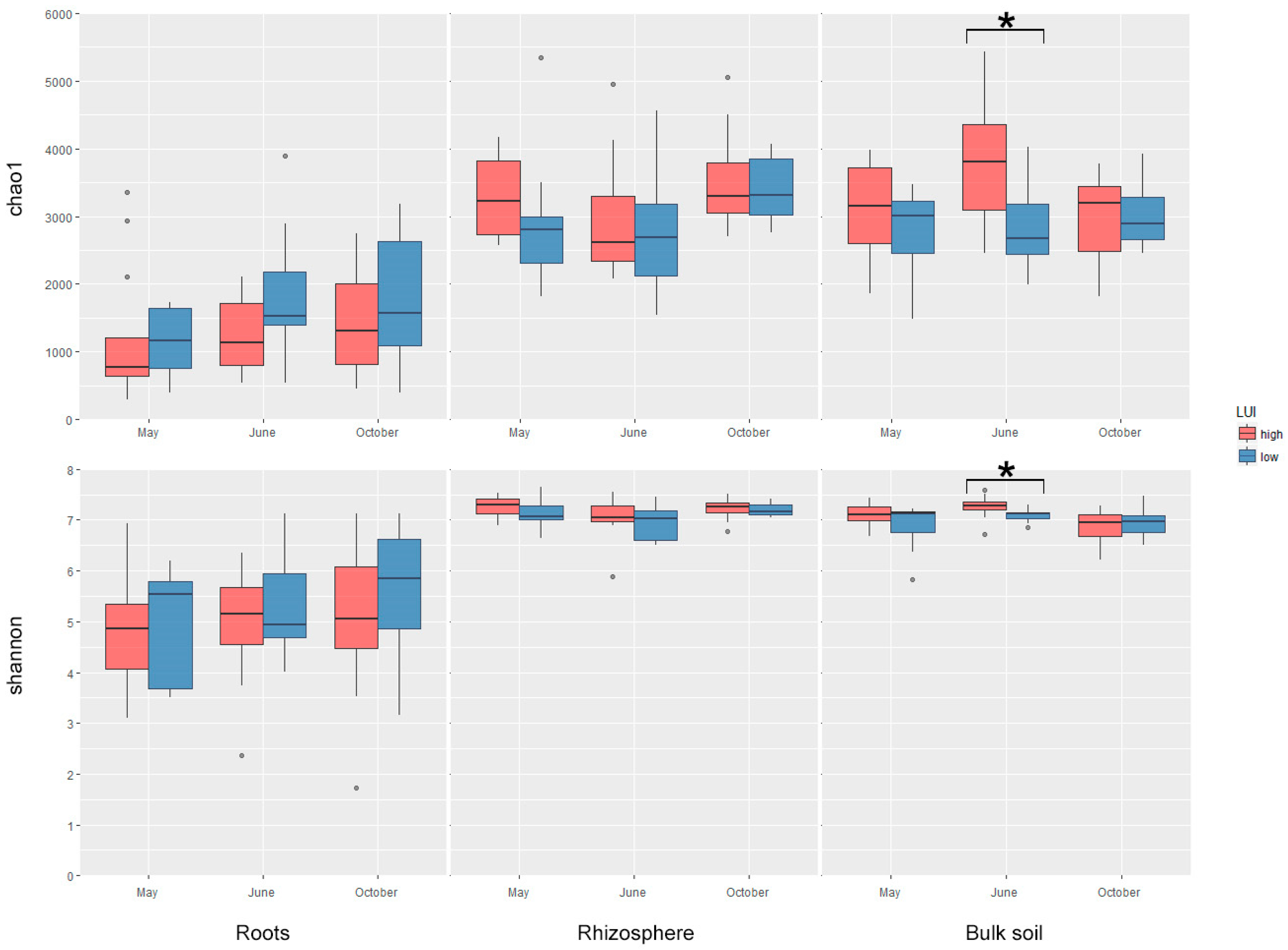
3.3. Characterization of Bacterial Diversity
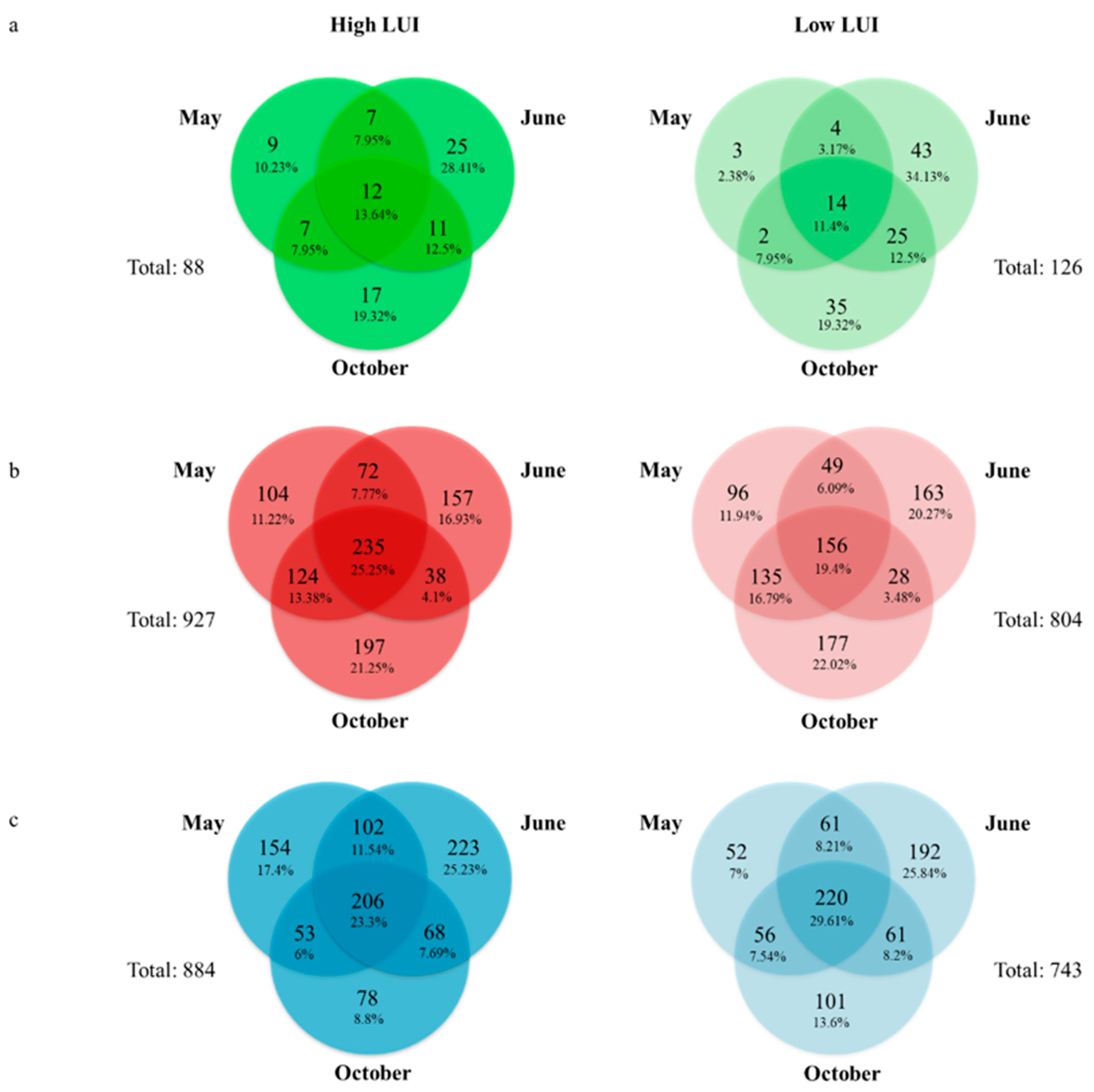
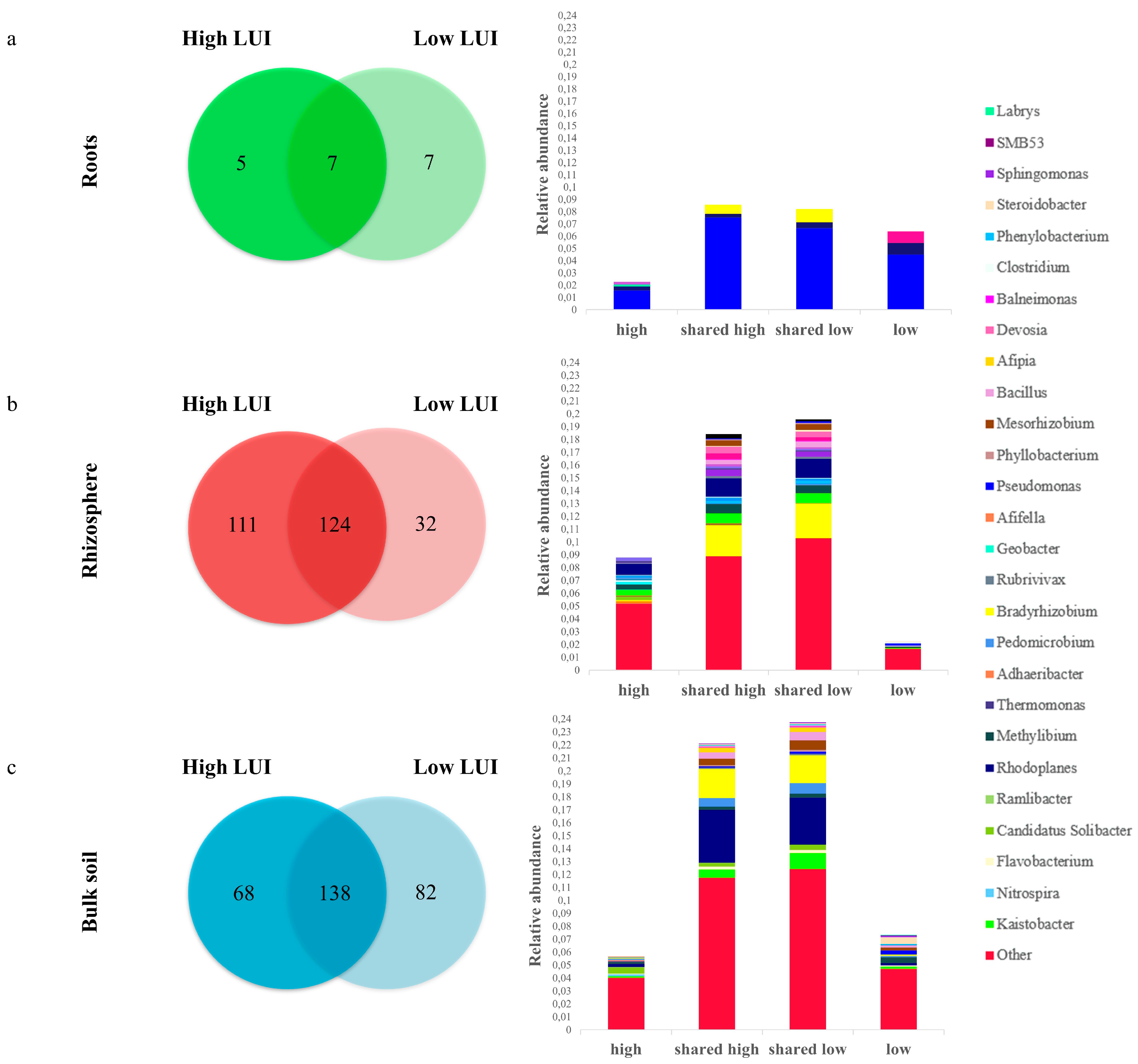
3.4. Definition of an Endophytic Core Microbiome for Dactylis glomerata L.
3.5. Definition of a Core Microbiome for the Rhizosphere of Dactylis glomerata L.
3.6. Definition of a Core Microbiome for the Bulk Soil of Dactylis glomerata L.
4. Discussion
4.1. The Role of LUI for Bacterial Diversity at the Plant–Soil Interface at Different Stages of Plant Development
4.2. The LUI Independent Bacterial Core Microbiome of D. glomerate
4.3. Microbial Variation of Core OTUs as Influenced by LUI
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martínez-Viveros, O.; Jorquera, M.A.; Crowley, D.E.; Gajardo, G.; Mora, M.L. Mechanisms and practical considerations involved in plant growth promotion by rhizobacteria. J. Soil. Sci. Plant. Nutr. 2010, 10, 293–319. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A.H.M. The rhizosphere microbiome and plant health. Trends Plant. Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Gaiero, J.; McCall, C.; Thompson, K.; Day, N.; Best, A.; Dunfield, K. Inside the root microbiome: Bacterial root endophytes and plant growth promotion. Am. J. Bot. 2013, 100. [Google Scholar] [CrossRef]
- Reinhold-Hurek, B.; Bünger, W.; Burbano, C.S.; Sabale, M.; Hurek, T. Roots shaping their microbiome: Global hotspots for microbial activity. Annu. Rev. Phytopathol. 2015, 53, 403–424. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.R.; James, E.K.; Poole, P.S. The plant microbiome. Genome. Biol. 2013, 14, 209. [Google Scholar] [CrossRef] [PubMed]
- Lugtenberg, B.; Kamilova, F. Plant-Growth-Promoting Rhizobacteria. Annu. Rev. Microbiol. 2009, 63, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Reinhold-Hurek, B.; Hurek, T. Living inside plants: Bacterial endophytes. Curr. Opin. Plant. Biol. 2011, 14, 435–443. [Google Scholar] [CrossRef]
- Truyens, S.; Weyens, N.; Cuypers, A.; Vangronsveld, J. Bacterial seed endophytes: Genera, vertical transmission and interaction with plants. Environ. Microbiol. Rep. 2015, 7, 40–50. [Google Scholar] [CrossRef]
- Helliwell, J.R.; Sturrock, C.J.; Miller, A.J.; Whalley, W.R.; Mooney, S.J. The role of plant species and soil condition in the structural development of the rhizosphere. Plant. Cell Environ. 2019, 42, 1974–1986. [Google Scholar] [CrossRef]
- Kowalchuk, G.A.; Buma, D.S.; de Boer, W.; Klinkhamer, P.G.L.; van Veen, J.A. Effects of above-ground plant species composition and diversity on the diversity of soil-borne microorganisms. Antonie Van Leeuwenhoek 2002, 81, 509. [Google Scholar] [CrossRef]
- Compant, S.; Reiter, B.; Sessitsch, A.; Nowak, J.; Clément, C.; Ait Barka, E. Endophytic Colonization of Vitis vinifera L. by Plant Growth-Promoting Bacterium Burkholderia sp. Strain PsJN. Appl. Environ. Microbiol. 2005, 71, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, N.; Sopory, S.K. Chemical signaling under abiotic stress environment in plants. Plant. Signal. Behav. 2008, 3, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Ernebjerg, M.; Kishony, R. Distinct Growth Strategies of Soil Bacteria as Revealed by Large-Scale Colony Tracking. Appl. Environ. Microbiol. 2012, 78, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Tardieu, F. Plant response to environmental conditions: Assessing potential production, water demand, and negative effects of water deficit. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Estendorfer, J.; Stempfhuber, B.; Haury, P.; Vestergaard, G.; Rillig, M.C.; Joshi, J.; Schröder, P.; Schloter, M. The Influence of Land Use Intensity on the Plant-Associated Microbiome of Dactylis glomerata L. Front. Plant. Sci. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.; Lebeis, S.; Herrera Paredes, S.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Rio, T.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef]
- Pfeiffer, S.; Mitter, B.; Oswald, A.; Schloter-Hai, B.; Schloter, M.; Declerck, S.; Sessitsch, A. Rhizosphere microbiomes of potato cultivated in the High Andes show stable and dynamic core microbiomes with different responses to plant development. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef]
- Bowen, J.L.; Kearns, P.J.; Byrnes, J.E.K.; Wigginton, S.; Allen, W.J.; Greenwood, M.; Tran, K.; Yu, J.; Cronin, J.T.; Meyerson, L.A. Lineage overwhelms environmental conditions in determining rhizosphere bacterial community structure in a cosmopolitan invasive plant. Nat. Commun. 2017, 8, 433. [Google Scholar] [CrossRef]
- Bluthgen, N.; Dormann, C.F.; Prati, D.; Klaus, V.H.; Kleinebecker, T.; Holzel, N.; Alt, F.; Boch, S.; Gockel, S.; Hemp, A.; et al. A quantitative index of land-use intensity in grasslands: Integrating mowing, grazing and fertilization. Basic Appl. Ecol. 2012, 13, 207–220. [Google Scholar] [CrossRef]
- Houba, V.J.G.; Temminghoff, E.J.M.; Gaikhorst, G.A.; van Vark, W. Soil analysis procedures using 0.01 M calcium chloride as extraction reagent. Commun. Soil. Sci. Plant. Anal. 2000, 31, 1299–1396. [Google Scholar] [CrossRef]
- Lueders, T.; Manefield, M.; Friedrich, M.W. Enhanced sensitivity of DNA- and rRNA-based stable isotope probing by fractionation and quantitative analysis of isopycnic centrifugation gradients. Environ. Microbiol. 2004, 6, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Dorn-In, S.; Bassitta, R.; Schwaiger, K.; Bauer, J.; Hölzel, C.S. Specific amplification of bacterial DNA by optimized so-called universal bacterial primers in samples rich of plant DNA. J. Microbiol. Methods 2015, 113, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS ONE 2011, 6, e17288. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric Estimation of the Number of Classes in a Population. Scand. Stat. Theory Appl. 1984, 11, 265–270. [Google Scholar]
- Southwood, T.R.E.; Henderson, P.A. Ecological Methods, 3rd ed.; Blackwell Science Ltd.: Hoboken, NJ, USA, 2000; p. 592. [Google Scholar]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and Qualitative β Diversity Measures Lead to Different Insights into Factors That Structure Microbial Communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef]
- Oksanen, F.J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.; O’Hara, R.B.; Simpson, G.; Solymos, P.; et al. Vegan: Community Ecology Package. Available online: https://cran.r-project.org/src/contrib/Archive/vegan/ (accessed on 10 August 2017).
- Calculate and Draw Custom Venn Diagrams. Available online: http://bioinformatics.psb.ugent.be/webtools/Venn/ (accessed on 15 January 2018).
- Garbeva, P.; van Elsas, J.D.; van Veen, J.A. Rhizosphere microbial community and its response to plant species and soil history. Plant. Soil. 2008, 302, 19–32. [Google Scholar] [CrossRef]
- Bevivino, A.; Paganin, P.; Bacci, G.; Florio, A.; Pellicer, M.S.; Papaleo, M.C.; Mengoni, A.; Ledda, L.; Fani, R.; Benedetti, A.; et al. Soil bacterial community response to differences in agricultural management along with seasonal changes in a Mediterranean region. PLoS ONE 2014, 9, e105515. [Google Scholar] [CrossRef] [PubMed]
- Grayston, S.J.; Wang, S.; Campbell, C.D.; Edwards, A.C. Selective influence of plant species on microbial diversity in the rhizosphere. Soil. Biol. Biochem. 1998, 30, 369–378. [Google Scholar] [CrossRef]
- Gottel, N.R.; Castro, H.F.; Kerley, M.; Yang, Z.; Pelletier, D.A.; Podar, M.; Karpinets, T.; Uberbacher, E.; Tuskan, G.A.; Vilgalys, R.; et al. Distinct microbial communities within the endosphere and rhizosphere of Populus deltoides roots across contrasting soil types. Appl. Environ. Microbiol. 2011, 77, 5934–5944. [Google Scholar] [CrossRef] [PubMed]
- Munier-Jolain, N.; Salon, C. Are the carbon costs of seed production related to the quantitative and qualitative performance? An appraisal of legumes and other crops. Plant. Cell Environ. 2005, 28, 1388–1395. [Google Scholar] [CrossRef]
- Berg, G.; Grube, M.; Schloter, M.; Smalla, K. Unraveling the plant microbiome: Looking back and future perspectives. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef]
- Lareen, A.; Burton, F.; Schäfer, P. Plant root-microbe communication in shaping root microbiomes. Plant. Mol. Biol. 2016, 90, 575–587. [Google Scholar] [CrossRef]
- Montes, C.; Altimira, F.; Canchignia, H.; Castro, Á.; Sánchez, E.; Miccono, M.; Tapia, E.; Sequeida, Á.; Valdés, J.; Tapia, P.; et al. A draft genome sequence of Pseudomonas veronii R4: A grapevine (Vitis vinifera L.) root-associated strain with high biocontrol potential. Stand. Genom. Sci. 2016, 11, 76. [Google Scholar] [CrossRef]
- Perrot-Rechenmann, C. Cellular responses to auxin: Division versus expansion. Cold Spring Harb. Perspect. Biol. 2010, 2, a001446. [Google Scholar] [CrossRef]
- Canchignia, H.; Altimira, F.; Montes, C.; Sánchez, E.; Tapia, E.; Miccono, M.; Espinoza, D.; Aguirre, C.; Seeger, M.; Prieto, H. Candidate nematicidal proteins in a new Pseudomonas veronii isolate identified by its antagonistic properties against Xiphinema index. J. Gen. Appl. Microbiol. 2017, 63, 11–21. [Google Scholar] [CrossRef]
- Viketoft, M.; Palmborg, C.; Sohlenius, B.; Huss-Danell, K.; Bengtsson, J. Plant species effects on soil nematode communities in experimental grasslands. Appl. Soil Ecol. 2005, 30, 90–103. [Google Scholar] [CrossRef]
- Oteino, N.; Lally, R.D.; Kiwanuka, S.; Lloyd, A.; Ryan, D.; Germaine, K.J.; Dowling, D.N. Plant growth promotion induced by phosphate solubilizing endophytic Pseudomonas isolates. Front. Microbiol. 2015, 6, 745. [Google Scholar] [CrossRef] [PubMed]
- Lodwig, E.M.; Hosie, A.H.F.; Bourdès, A.; Findlay, K.; Allaway, D.; Karunakaran, R.; Downie, J.A.; Poole, P.S. Amino-acid cycling drives nitrogen fixation in the legume–Rhizobium symbiosis. Nature 2003, 422, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Aoki, S.; Kawaguchi, M. Evolutionary Dynamics of Nitrogen Fixation in the Legume–Rhizobia Symbiosis. PLoS ONE 2014, 9, e93670. [Google Scholar] [CrossRef]
- Chi, F.; Shen, S.-H.; Cheng, H.-P.; Jing, Y.-X.; Yanni, Y.G.; Dazzo, F.B. Ascending migration of endophytic rhizobia, from roots to leaves, inside rice plants and assessment of benefits to rice growth physiology. Appl. Environ. Microbiol. 2005, 71, 7271–7278. [Google Scholar] [CrossRef] [PubMed]
- Egamberdieva, D.; Wirth, S.J.; Alqarawi, A.A.; Abd_Allah, E.F.; Hashem, A. Phytohormones and Beneficial Microbes: Essential Components for Plants to Balance Stress and Fitness. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Pfitzner, B.; Schmid, M.; Simões-Araújo, J.L.; Reis, V.M.; Pereira, W.; Ormeño-Orrillo, E.; Hai, B.; Hofmann, A.; Schloter, M.; et al. Molecular characterisation of the diazotrophic bacterial community in uninoculated and inoculated field-grown sugarcane (Saccharum sp.). Plant. Soil 2012, 356, 83–99. [Google Scholar] [CrossRef]
- Nakatsu, C.H.; Hristova, K.; Hanada, S.; Meng, X.-Y.; Hanson, J.R.; Scow, K.M.; Kamagata, Y. Methylibium petroleiphilum gen. nov., sp. nov., a novel methyl tert-butyl ether-degrading methylotroph of the Betaproteobacteria. Int. J. Syst. Evol. Microbiol. 2006, 56, 983–989. [Google Scholar] [CrossRef]
- Mao, Y.; Li, X.; Smyth, E.M.; Yannarell, A.C.; Mackie, R.I. Enrichment of specific bacterial and eukaryotic microbes in the rhizosphere of switchgrass (Panicum virgatum L.) through root exudates. Environ. Microbiol. Rep. 2014, 6, 293–306. [Google Scholar] [CrossRef]
- Rouws, L.; Leite, J.; Feitosa de Matos, G.; Zilli, J.; Reed Rodrigues Coelho, M.; Xavier, G.; Fischer, D.; Hartmann, A.; Reis, V.; Baldani, J. Endophytic Bradyrhizobium spp. isolates from sugarcane obtained through different culture strategies. Environ. Microbiol. Rep. 2013, 6. [Google Scholar] [CrossRef]
- Gkarmiri, K.; Mahmood, S.; Ekblad, A.; Alström, S.; Högberg, N.; Finlay, R. Identifying the Active Microbiome Associated with Roots and Rhizosphere Soil of Oilseed Rape. Appl. Environ. Microbiol. 2017, 83, e01938-17. [Google Scholar] [CrossRef]
- Hiraishi, A.; Ueda, Y. Rhodoplanes gen. nov., a New Genus of Phototrophic Bacteria Including Rhodopseudomonas rosea as Rhodoplanes roseus comb. nov. and Rhodoplanes elegans sp. nov. Int. J. Syst. Evol. Microbiol. 1994, 44, 665–673. [Google Scholar] [CrossRef]
- Buckley, D.H.; Huangyutitham, V.; Hsu, S.-F.; Nelson, T.A. Stable Isotope Probing with 15N2 Reveals Novel Noncultivated Diazotrophs in Soil. Appl. Environ. Microbiol. 2007, 73, 3196–3204. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, S.; Jiang, Q.; Bai, Y.; Shen, G.; Li, S.; Ding, W. Using community analysis to explore bacterial indicators for disease suppression of tobacco bacterial wilt. Sci. Rep. 2016, 6, 36773. [Google Scholar] [CrossRef] [PubMed]
- Chimwamurombe, P.M.; Grönemeyer, J.L.; Reinhold-Hurek, B. Isolation and characterization of culturable seed-associated bacterial endophytes from gnotobiotically grown Marama bean seedlings. FEMS Microbiol. Ecol. 2016, 92, fiw083. [Google Scholar] [CrossRef]
- Campisano, A.; Antonielli, L.; Pancher, M.; Yousaf, S.; Pindo, M.; Pertot, I. Bacterial Endophytic Communities in the Grapevine Depend on Pest Management. PLoS ONE 2014, 9, e112763. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.A.; Kalyuzhnaya, M.G.; Noyes, E.; Lara, J.C.; Lidstrom, M.E.; Chistoserdova, L. Labrys methylaminiphilus sp. nov., a novel facultatively methylotrophic bacterium from a freshwater lake sediment. Int. J. Syst. Evol. Microbiol. 2005, 55, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.; De Marco, P.; Duque, A.; Pacheco, C.; Janssen, D.; Castro, P. Labrys portucalensis sp. nov., a fluorobenzene-degrading bacterium isolated from an industrially contaminated sediment in northern Portugal. Int. J. Syst. Evol. Microbiol. 2008, 58, 692–698. [Google Scholar] [CrossRef]
- Nguyen, N.-L.; Kim, Y.-J.; Hoang, V.-A.; Kang, J.-P.; Wang, C.; Zhang, J.; Kang, C.-H.; Yang, D.-C. Labrys soli sp. nov., isolated from the rhizosphere of ginseng. Int. J. Syst. Evol. Microbiol. 2015, 65, 3913–3919. [Google Scholar] [CrossRef]
- Passari, A.K.; Mishra, V.K.; Leo, V.V.; Gupta, V.K.; Singh, B.P. Phytohormone production endowed with antagonistic potential and plant growth promoting abilities of culturable endophytic bacteria isolated from Clerodendrum colebrookianum Walp. Microbiol. Res. 2016, 193, 57–73. [Google Scholar] [CrossRef]
- Chihaoui, S.A.; Trabelsi, D.; Jdey, A.; Mhadhbi, H.; Mhamdi, R. Inoculation of Phaseolus vulgaris with the nodule-endophyte Agrobacterium sp. 10C2 affects richness and structure of rhizosphere bacterial communities and enhances nodulation and growth. Arch. Microbiol. 2015, 197, 805–813. [Google Scholar] [CrossRef]
- Bosmans, L.; Moerkens, R.; Wittemans, L.; De Mot, R.; Rediers, H.; Lievens, B. Rhizogenic agrobacteria in hydroponic crops: Epidemics, diagnostics and control. Plant. Pathol. 2017, 66, 1043–1053. [Google Scholar] [CrossRef]
- Pereira, S.I.; Castro, P.M. Diversity and characterization of culturable bacterial endophytes from Zea mays and their potential as plant growth-promoting agents in metal-degraded soils. Environ. Sci. Pollut. Res. 2014, 21, 14110–14123. [Google Scholar] [CrossRef]
- Leadbetter, J.R.; Greenberg, E.P. Metabolism of Acyl-Homoserine Lactone Quorum-Sensing Signals by Variovorax paradoxus. J. Bacteriol. 2000, 182, 6921–6926. [Google Scholar] [CrossRef] [PubMed]
- Han, J.-I.; Choi, H.-K.; Lee, S.-W.; Orwin, P.M.; Kim, J.; LaRoe, S.L.; Kim, T.-g.; O’Neil, J.; Leadbetter, J.R.; Lee, S.Y.; et al. Complete Genome Sequence of the Metabolically Versatile Plant Growth-Promoting Endophyte Variovorax paradoxus S110. J. Bacteriol. 2011, 193, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.Y.; Fulthorpe, R. Seasonal variation of bacterial endophytes in urban trees. Front. Microbiol. 2015, 6, 427. [Google Scholar] [CrossRef]
- Khan, A.L.; Waqas, M.; Asaf, S.; Kamran, M.; Shahzad, R.; Bilal, S.; Khan, M.A.; Kang, S.-M.; Kim, Y.-H.; Yun, B.-W.; et al. Plant growth-promoting endophyte Sphingomonas sp. LK11 alleviates salinity stress in Solanum pimpinellifolium. Environ. Exp. Bot. 2017, 133, 58–69. [Google Scholar] [CrossRef]
- Correa-Galeote, D.; Bedmar, E.J.; Arone, G.J. Maize Endophytic Bacterial Diversity as Affected by Soil Cultivation History. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.A.; Jin, Z.; Youngblut, N.; Wallace, J.G.; Sutter, J.; Zhang, W.; González-Peña, A.; Peiffer, J.; Koren, O.; Shi, Q.; et al. Large-scale replicated field study of maize rhizosphere identifies heritable microbes. Proc. Natl. Acad. Sci. USA 2018, 115, 7368–7373. [Google Scholar] [CrossRef]
- Dohrmann, A.; Küting, M.; Jünemann, S.; Jaenicke, S.; Schlueter, A.; Tebbe, C. Importance of rare taxa for bacterial diversity in the rhizosphere of Bt-and conventional maize varieties. ISME J. 2012, 7. [Google Scholar] [CrossRef]
- Egamberdiyeva, D.; Höflich, G. Influence of growth-promoting bacteria on the growth of wheat in different soils and temperatures. Soil. Biol. Biochem. 2003, 35, 973–978. [Google Scholar] [CrossRef]
- Fernández-González, A.J.; Martínez-Hidalgo, P.; Cobo-Díaz, J.F.; Villadas, P.J.; Martínez-Molina, E.; Toro, N.; Tringe, S.G.; Fernández-López, M. The rhizosphere microbiome of burned holm-oak: Potential role of the genus Arthrobacter in the recovery of burned soils. Sci. Rep. 2017, 7, 6008. [Google Scholar] [CrossRef] [PubMed]
- Madigan, M.; Cox, S.S.; Stegeman, R.A. Nitrogen fixation and nitrogenase activities in members of the family Rhodospirillaceae. J. Bacteriol. 1984, 157, 73–78. [Google Scholar] [CrossRef]
- Laranjo, M.; Alexandre, A.; Oliveira, S. Legume growth-promoting rhizobia: An overview on the Mesorhizobium genus. Microbiol. Res. 2014, 169, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Hardoim, P.R.; van Overbeek, L.S.; Berg, G.; Pirttilä, A.M.; Compant, S.; Campisano, A.; Döring, M.; Sessitsch, A. The Hidden World within Plants: Ecological and Evolutionary Considerations for Defining Functioning of Microbial Endophytes. Microbiol. Mol. Biol. Rev. MMBR 2015, 79, 293–320. [Google Scholar] [CrossRef] [PubMed]
- Dreccer, M.F.; Chapman, S.C.; Rattey, A.R.; Neal, J.; Song, Y.; Christopher, J.J.; Reynolds, M. Developmental and growth controls of tillering and water-soluble carbohydrate accumulation in contrasting wheat (Triticum aestivum L.) genotypes: Can we dissect them? J. Exp. Bot. 2013, 64, 143–160. [Google Scholar] [CrossRef]
- Fitzpatrick, C.R.; Salas-González, I.; Conway, J.M.; Finkel, O.M.; Gilbert, S.; Russ, D.; Pereira Lima Teixeira, P.J.; Dangl, J.L. The Plant Microbiome: From Ecology to Reduction and Beyond. Annu. Rev. Microbiol. 2020, 74, 81–100. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Estendorfer, J.; Stempfhuber, B.; Vestergaard, G.; Schulz, S.; Rillig, M.C.; Joshi, J.; Schröder, P.; Schloter, M. Definition of Core Bacterial Taxa in Different Root Compartments of Dactylis glomerata, Grown in Soil under Different Levels of Land Use Intensity. Diversity 2020, 12, 392. https://doi.org/10.3390/d12100392
Estendorfer J, Stempfhuber B, Vestergaard G, Schulz S, Rillig MC, Joshi J, Schröder P, Schloter M. Definition of Core Bacterial Taxa in Different Root Compartments of Dactylis glomerata, Grown in Soil under Different Levels of Land Use Intensity. Diversity. 2020; 12(10):392. https://doi.org/10.3390/d12100392
Chicago/Turabian StyleEstendorfer, Jennifer, Barbara Stempfhuber, Gisle Vestergaard, Stefanie Schulz, Matthias C. Rillig, Jasmin Joshi, Peter Schröder, and Michael Schloter. 2020. "Definition of Core Bacterial Taxa in Different Root Compartments of Dactylis glomerata, Grown in Soil under Different Levels of Land Use Intensity" Diversity 12, no. 10: 392. https://doi.org/10.3390/d12100392
APA StyleEstendorfer, J., Stempfhuber, B., Vestergaard, G., Schulz, S., Rillig, M. C., Joshi, J., Schröder, P., & Schloter, M. (2020). Definition of Core Bacterial Taxa in Different Root Compartments of Dactylis glomerata, Grown in Soil under Different Levels of Land Use Intensity. Diversity, 12(10), 392. https://doi.org/10.3390/d12100392