A Multireference-Based Whole Genome Assembly for the Obligate Ant-Following Antbird, Rhegmatorhina melanosticta (Thamnophilidae)


Abstract
1. Introduction
2. Methods
2.1. Genome Sequencing and De Novo Assembly
2.2. Single Reference Assisted Chromosome Level Assembly
2.3. Multiple Reference Assisted Chromosome Level Assembly
2.4. Evaluation of Genome Completeness
2.5. Genotyping-by-Sequencing (GBS) Reference Mapping
3. Results
3.1. De Novo Assembly
3.2. Reference-Based Assembly
3.3. Genome Completeness
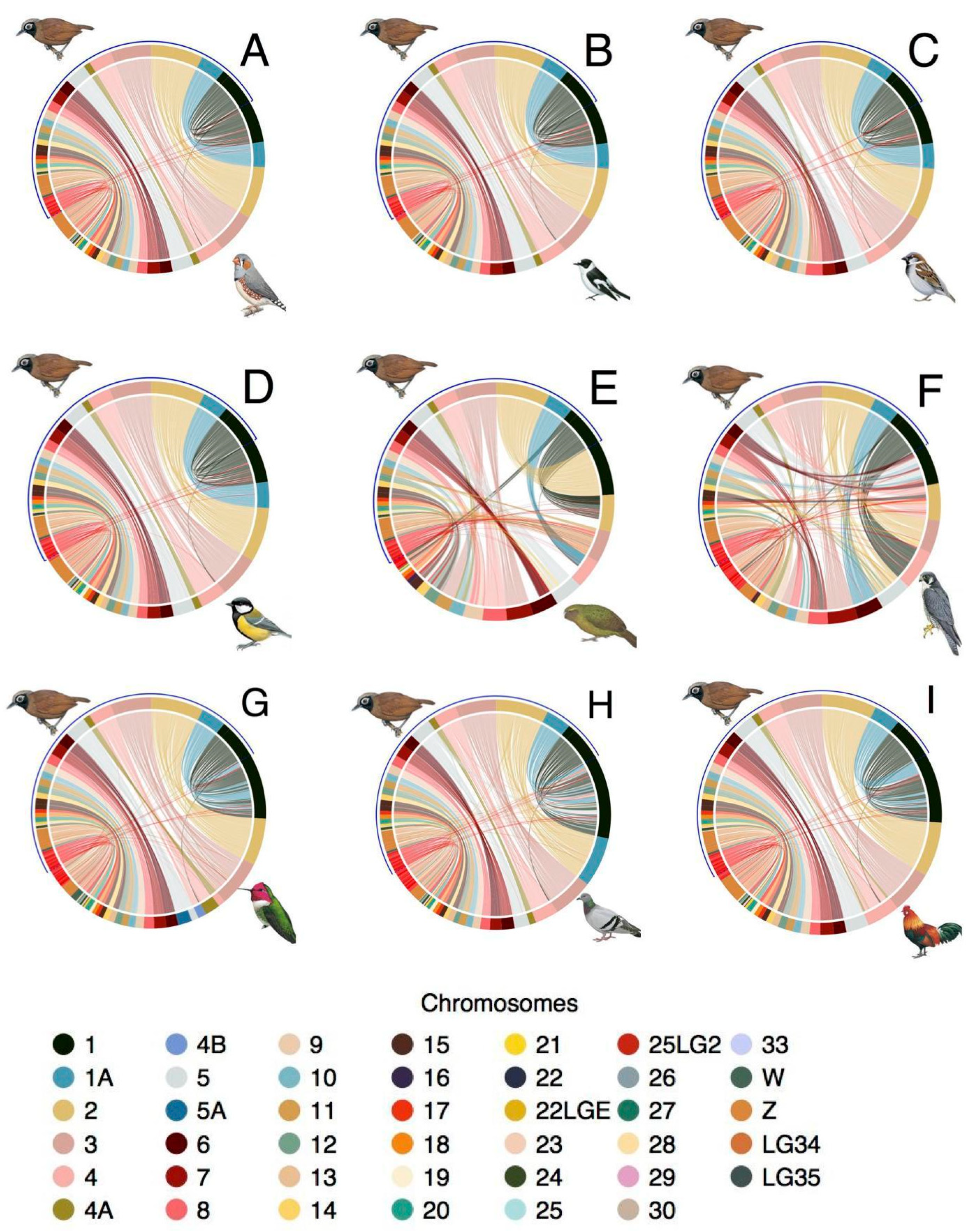
3.4. Synteny
3.5. GBS Reference Mapping
4. Discussion
4.1. General Genome Structure, Contiguity and Content
4.2. Analysis of Synteny
4.3. Linked-Read Genome Applicability in Comparative Phylogenomics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Ellegren, H. Genome sequencing and population genomics in non-model organisms. Trends Ecol. Evol. 2014, 29, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G. Genomics: Bird sequencing project takes off. Nature 2015, 522, 34. [Google Scholar] [CrossRef] [PubMed]
- Genome 10K Community of Scientists. Genome 10K: A proposal to obtain whole-genome sequence for 10,000 vertebrate species. J. Hered. 2009, 100, 659–674. [Google Scholar] [CrossRef] [PubMed]
- Koepfli, K.-P.; Paten, B.; the Genome 10K Community of Scientists; O’Brien, S.J. The Genome 10K Project: A way forward. Annu. Rev. Anim. Biosci. 2015, 3, 57–111. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.W.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Phylogenomic analyses data of the avian phylogenomics project. Gigascience 2015, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, C.; Li, Q.; Li, B.; Larkin, D.M.; Lee, C.; Storz, J.F.; Antunes, A.; Greenwold, M.J.; Meredith, R.W.; et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 2014, 346, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.W.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Prum, R.O.; Berv, J.S.; Dornburg, A.; Field, D.J.; Townsend, J.P.; Lemmon, E.M.; Lemmon, A.R. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature 2015, 526, 569–573. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.E.; Harvey, M.G.; Faircloth, B.C.; Crawford, N.G.; Glenn, T.C.; Brumfield, R.T. A phylogeny of birds based on over 1,500 loci collected by target enrichment and high-throughput sequencing. PLoS ONE 2013, 8, e54848. [Google Scholar] [CrossRef]
- Toews, D.P.L.; Taylor, S.A.; Vallender, R.; Brelsford, A.; Butcher, B.G.; Messer, P.W.; et al. Plumage Genes and Little Else Distinguish the Genomes of Hybridizing Warblers. Curr. Biol. 2016, 26, 2313–2318. [Google Scholar] [CrossRef]
- Harvey, M.G.; Aleixo, A.; Ribas, C.C.; Brumfield, R.T. Habitat Association Predicts Genetic Diversity and Population Divergence in Amazonian Birds. Am. Nat. 2017, 190, 631–648. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.T.; Harvey, M.G.; Faircloth, B.C.; Glenn, T.C. Target capture and massively parallel sequencing of ultraconserved elements for comparative studies at shallow evolutionary time scales. Syst. Biol. 2013, 63, 83–95. Available online: https://academic.oup.com/sysbio/article-abstract/63/1/83/1689074 (accessed on 15 August 2019). [CrossRef] [PubMed]
- Raposo do Amaral, F.; Maldonado-Coelho, M.; Aleixo, A.; Luna, L.W.; Rêgo, P.S.D.; Araripe, J.; Souza, T.O.; Silva, W.A.G.; Thom, G. Recent chapters of Neotropical history overlooked in phylogeography: Shallow divergence explains phenotype genotype uncoupling in Antilophia manakins. Mol. Ecol. 2018, 27, 4108–4120. [Google Scholar] [CrossRef] [PubMed]
- Oswald, J.A.; Harvey, M.G.; Remsen, R.C.; Foxworth, D.U.; Dittmann, D.L.; Cardiff, S.W.; Brumfield, R.T. Evolutionary dynamics of hybridization introgression following the recent colonization of Glossy Ibis (Aves: Plegadis falcinellus) into the New World. Mol. Ecol. 2019, 28, 1675–1691. [Google Scholar] [CrossRef] [PubMed]
- Oswald, J.A.; Overcast, I.; Mauck, W.M.; Andersen, M.J.; Smith, B.T. Isolation with asymmetric gene flow during the nonsynchronous divergence of dry forest birds. Mol Ecol. 2017, 26, 1386–1400. [Google Scholar] [CrossRef] [PubMed]
- Nadachowska-Brzyska, K.; Li, C.; Smeds, L.; Zhang, G.; Ellegren, H. Temporal Dynamics of Avian Populations during Pleistocene Revealed by Whole-Genome Sequences. Curr. Biol. 2015, 25, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Musher, L.J.; Ferreira, M.; Auerbach, A.L.; McKay, J.; Cracraft, J. Why is Amazonia a “source” of biodiversity? Climate-mediated dispersal and synchronous speciation across the Andes in an avian group (Tityrinae). Proc. R. Soc. B. 2019, 286, 20182343. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, C.H.; Field, D.J.; Ksepka, D.T.; Barker, F.K.; Aleixo, A.; Andersen, M.J.; Alström, P.; Benz, B.W.; Braun, E.L.; Braun, M.J.; et al. Earth history and the passerine superradiation. Proc. Natl. Acad. Sci. USA 2019, 116, 7916–7925. [Google Scholar] [CrossRef]
- Nam, K.; Mugal, C.; Nabholz, B.; Schielzeth, H.; Wolf, J.B.W.; Backström, N.; Künstner, A.; Balakrishnan, C.N.; Heger, A.; Ponting, C.P.; et al. Molecular evolution of genes in avian genomes. Genome Biol. 2010, 11, R68. [Google Scholar] [CrossRef] [PubMed]
- Ellegren, H.; Smeds, L.; Burri, R.; Olason, P.I.; Backström, N.; Kawakami, T.; Künstner, A.; Mäkinen, H.; Nadachowska-Brzyska, K.; Qvarnström, A.; et al. The genomic landscape of species divergence in Ficedula flycatchers. Nature 2012, 491, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Runemark, A.; Trier, C.N.; Eroukhmanoff, F.; Hermansen, J.S.; Matschiner, M.; Ravinet, M.; Elgvin, T.O.; Sætre, G.-P. Variation and constraints in hybrid genome formation. Nat. Ecol. Evol. 2018, 2, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.E.; Milá, B.; Toews, D.P.L.; Brelsford, A.; Kenyon, H.L.; Porter, A.N.; Grossen, C.; Delmore, K.E.; Alcaide, M.; Irwin, J.H. A comparison of genomic islands of differentiation across three young avian species pairs. Mol. Ecol. 2018, 27, 4839–4855. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, M.; Scordato, E.S.C.; Price, T.D.; Irwin, D.E. Genomic divergence in a ring species complex. Nature 2014, 511, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Hron, T.; Pajer, P.; Pačes, J.; Bartůněk, P.; Elleder, D. Hidden genes in birds. Genome biol. 2015, 16, 164. [Google Scholar] [CrossRef] [PubMed]
- Botero-Castro, F.; Figuet, E.; Tilak, M.-K.; Nabholz, B.; Galtier, N. Avian Genomes Revisited: Hidden Genes Uncovered and the Rates versus Traits Paradox in Birds. Mol. Biol. Evol. 2017, 34, 3123–3131. [Google Scholar] [CrossRef] [PubMed]
- Tigano, A.; Sackton, T.B.; Friesen, V.L. Assembly and RNA-free annotation of highly heterozygous genomes: The case of the thick-billed murre (Uria lomvia). Mol. Ecol. Resour. 2018, 18, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Sotero-Caio, C.G.; Platt, R.N.; Suh, A.; Ray, D.A. Evolution and Diversity of Transposable Elements in Vertebrate Genomes. Genome Biol. Evol. 2017, 9, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Lien, S.; Koop, B.F.; Sandve, S.R.; Miller, J.R.; Kent, M.P.; Nome, T.; Hvidsten, T.R.; Leong, J.S.; Minkley, D.R.; Zimin, A.; et al. The Atlantic salmon genome provides insights into rediploidization. Nature 2016, 533, 200–205. [Google Scholar] [CrossRef]
- English, A.C.; Richards, S.; Han, Y.; Wang, M.; Vee, V.; Qu, J.; Qin, X.; Muzny, D.M.; Reid, J.G.; Worley, K.C.; et al. Mind the gap: Upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 2012, 7, e47768. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Korbel, J.O.; Urban, A.E.; Affourtit, J.P.; Godwin, B.; Grubert, F.; Simons, J.F.; Kim, P.M.; Palejev, D.; Carriero, N.J.; Du, L.; et al. Paired-end mapping reveals extensive structural variation in the human genome. Science 2007, 318, 420–426. [Google Scholar] [CrossRef]
- Myers, E.W.; Sutton, G.G.; Delcher, A.L.; Dew, I.M.; Fasulo, D.P.; Flanigan, M.J.; Kravitz, S.A.; Mobarry, C.M.; Reinert, K.H.J.; Remington, K.A.; et al. A whole-genome assembly of Drosophila. Science 2000, 287, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- Cheung, V.G.; Nowak, N.; Jang, W.; Kirsch, I.R.; Zhao, S.; Chen, X.N.; Furey, T.S.; Kim, U.J.; Kuo, W.L.; Olivier, M. Integration of cytogenetic landmarks into the draft sequence of the human genome. Nature 2001, 409, 953–958. [Google Scholar] [PubMed]
- Korlach, J.; Gedman, G.; Kingan, S.B.; Chin, C.-S.; Howard, J.T.; Audet, J.-N.; Cantin, L.; Jarvis, E.D. De novo PacBio long-read and phased avian genome assemblies correct and add to reference genes generated with intermediate and short reads. Gigascience 2017, 6, 1–16. [Google Scholar] [CrossRef]
- Ozerov, M.Y.; Ahmad, F.; Gross, R.; Pukk, L.; Kahar, S.; Kisand, V.; Vasemägi, A. Highly Continuous Genome Assembly of Eurasian Perch (Perca fluviatilis) Using Linked-Read Sequencing. G3: Genes Genomes Genet. 2018, 8, 3737–3743. [Google Scholar] [CrossRef] [PubMed]
- Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.B. Direct determination of diploid genome sequences. Genome Res. 2017, 27, 757–767. [Google Scholar] [CrossRef]
- Sohn, J.; Nam, J.W. The present and future of de novo whole-genome assembly. Brief. Bioinform. 2016. Available online: https://academic.oup.com/bib/article-abstract/19/1/23/2339783 (accessed on 15 August 2019).
- Sedlazeck, F.J.; Lee, H.; Darby, C.A.; Schatz, M.C. Piercing the dark matter: Bioinformatics of long-range sequencing and mapping [Internet]. Nat. Rev. Genet. 2018, 329–346. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Armstrong, J.; Raney, B.J.; Streeter, I.; Dunn, M.; Yang, F.; Odom, D.; Flicek, P.; Keane, T.M.; Thybert, D.; et al. Chromosome assembly of large and complex genomes using multiple references. Genome Res. 2018, 28, 1720–1732. [Google Scholar] [CrossRef]
- Tamazian, G.; Dobrynin, P.; Krasheninnikova, K.; Komissarov, A.; Koepfli, K.-P.; O’Brien, S.J. Chromosomer: A reference-based genome arrangement tool for producing draft chromosome sequences. Gigascience 2016, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Larkin, D.M.; Cai, Q.; Asan Zhang, Y.; Ge, R.-L.; Auvil, L.; Capitanu, B.; Zhang, G.; Lewin, H.A.; Ma, J. Reference-assisted chromosome assembly. Proc. Natl. Acad. Sci. USA 2013, 110, 1785–1790. [Google Scholar] [CrossRef]
- Marcondes, R.S.; Brumfield, R.T. Fifty shades of brown: Macroevolution of plumage brightness in the Furnariida, a large clade of drab Neotropical passerines. Evolution 2019, 73, 704–719. [Google Scholar] [CrossRef]
- Seeholzer, G.F.; Claramunt, S.; Brumfield, R.T. Niche evolution and diversification in a Neotropical radiation of birds (Aves: Furnariidae). Evolution 2017, 71, 702–715. [Google Scholar] [CrossRef] [PubMed]
- Raikow, R.J. Why are there so many kinds of passerine birds? Syst. Zool. 1986, 35, 255–259. [Google Scholar] [CrossRef]
- Derryberry, E.P.; Claramunt, S.; Darryberry, G.; Chesser, R.T.; Cracraft, J.; Aleixo, A.; Péres-Emán, J.; Remnsen, J.V.; Brumfield, R.T. Lineage diversification and morphological evolution in a large-scale continental radiation: The Neotropical ovenbirds and woodcreepers (Aves: Furnariidae). Evolution. 2011. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1558-5646.2011.01374.x (accessed on 22 August 2019).
- Isler, M.L.; Isler, P.R.; Whitney, B.M. Species Limits in Antbirds (Thamnophilidae): The Warbling Antbird (Hypocnemis Cantator) Complex. Auk 2007, 124, 11–28. [Google Scholar] [CrossRef]
- Willis, E.O. Taxonomy and behavior of Pale-faced Antbirds. Auk 1968, 85, 253–264. [Google Scholar] [CrossRef]
- Moyle, R.G.; Chesser, R.T.; Brumfield, R.T.; Tello, J.G. Phylogeny and phylogenetic classification of the antbirds, ovenbirds, woodcreepers, and allies (Aves: Passeriformes: Infraorder Furnariides). Cladistics. 2009. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1096-0031.2009.00259.x (accessed on 22 August 2019).
- Willis, E.O. On the behavior of five species of Rhegmatorhina, ant-following antbirds of the Amazon basin. Wilson Bull. 1969, 81, 363–395. [Google Scholar]
- Chaves-Campos, J. Ant colony tracking in the obligate army ant-following antbird Phaenostictus mcleannani. J. Ornithol. 2011, 152, 497–504. [Google Scholar] [CrossRef]
- Aleixo, A.; Burlamaqui, T.C.T.; Schneider, M.P.C.; Gonçalves, E.C. Molecular systematics and plumage evolution in the monotypic obligate army-ant-following genus Skutchia (Thamnophilidae). Condor 2009, 111, 382–387. [Google Scholar] [CrossRef]
- Ribas, C.C.; Aleixo, A.; Gubili, C.; d’Horta, F.M. Biogeography and diversification of Rhegmatorhina (Aves: Thamnophilidae): Implications for the evolution of Amazonian landscapes during the Quaternary. J. Biogeography. 2018. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1111/jbi.13169 (accessed on 22 August 2019).
- Isler, M.L.; Bravo, G.A.; Brumfield, R.T. Systematics of the obligate ant-following clade of antbirds (Aves: Passeriformes: Thamnophilidae). Wilson J. Ornithol. 2014, 126, 635–648. [Google Scholar] [CrossRef]
- Pulido-Santacruz, P.; Aleixo, A.; Weir, J.T. Morphologically cryptic Amazonian bird species pairs exhibit strong postzygotic reproductive isolation. Proc. Biol. Sci. 2018, 285. [Google Scholar] [CrossRef] [PubMed]
- Hackett, S.J. Phylogenetic and biogeographic relationships in the Neotropical genus Gymnopithys (Formicariidae). Wilson Bull. 1993, 105, 301–315. [Google Scholar]
- Silva, S.M.; Peterson, A.T.; Carneiro, L.; Burlamaqui, T.C.T.; Ribas, C.C.; Sousa-Neves, T.; Miranda, L.S.; Fernandes, A.F.; d’Horta, F.; Araújo-Silva, L.E.; et al. A dynamic continental moisture gradient drove Amazonian bird diversification. Sci. Adv. 2019, 5, eaat5752. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Schnable, J.C.; Yeh, C.-T.; Wu, L.; Liu, C.; Hu, H.-C.; DAlgrad, C.L.; Sarkar, S.; Schnable, P.S. Linked read technology for assembling large complex and polyploid genomes. BMC Genom. 2018, 19, 651. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.Y.; Lau, B.T.; Schnall-Levin, M.; Jarosz, M.; Bell, J.M.; Hindson, C.M.; Kyriazopoulou-Panagiotopoulou, S.; Masquelier, D.A.; Merrill, L.; Terry, J.M. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 2016, 34, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.; Garcia, S.; Barrio, A.M.; Belhocine, K.; Bernate, J.; Bharadwaj, R.; Bjornson, K.; Catalanotti, C.; Delaney, J.; Fehr, A.; et al. Resolving the full spectrum of human genome variation using Linked-Reads. Genome Res. 2019, 29, 635–645. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Ellegren, H. The Evolutionary Genomics of Birds. Annu. Rev. Ecol. Evol. Syst. 2013, 44, 239–259. [Google Scholar] [CrossRef]
- Sohn, J.-I.; Nam, K.; Hong, H.; Kim, J.-M.; Lim, D.; Lee, K.-T.; Do, Y.J.; Cho, C.Y.; Kim, N.; Chai, H.H.; et al. Whole genome and transcriptome maps of the entirely black native Korean chicken breed Yeonsan Ogye. Gigascience 2018, 7. [Google Scholar] [CrossRef]
- Damas, J.; O’Connor, R.E.; Griffin, D.K.; Larkin, D.M. Avian Chromosomal Evolution. In Avian Genomics in Ecology and Evolution: From the Lab into the Wild; Kraus, R.H.S., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 69–92. [Google Scholar]
- Laine, V.N.; Gossmann, T.I.; Schachtschneider, K.M.; Garroway, C.J.; Madsen, O.; Verhoeven, K.J.F.; de Jager, V.; Megens, H.J.; Warren, W.C.; Minx, P.; et al. Evolutionary signals of selection on cognition from the great tit genome and methylome. Nat. Commun. 2016, 7, 10474. [Google Scholar] [CrossRef]
- Elgvin, T.O.; Trier, C.N.; Tørresen, O.K.; Hagen, I.J.; Lien, S.; Nederbragt, A.J.; Ravinet, M.; Jensen, H.; Saetre, G.P. The genomic mosaicism of hybrid speciation. Sci. Adv. 2017, 3, e1602996. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Smeds, L.; Backström, N.; Husby, A.; Qvarnström, A.; Mugal, C.F.; Olason, P.; Ellegren, H. A high-density linkage map enables a second-generation collared flycatcher genome assembly and reveals the patterns of avian recombination rate variation and chromosomal evolution. Mol. Ecol. 2014, 23, 4035–4058. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Smeds, L.; Warmuth, V.; Bolivar, P.; Uebbing, S.; Burri, R.; Suh, A.; NAter, A.; Bureš, S.; Garamszegi, L.Z.; Hogner, S.; et al. Evolutionary analysis of the female-specific avian W chromosome. Nat. Commun. 2015, 6, 7330. [Google Scholar] [CrossRef] [PubMed]
- Bellott, D.W.; Skaletsky, H.; Cho, T.-J.; Brown, L.; Locke, D.; Chen, N.; Galkina, S.; Pyntikova, T.; Koutseva, N.; Graves, T.; et al. Avian W and mammalian Y chromosomes convergently retained dosage-sensitive regulators. Nat. Genet. 2017, 49, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Paten, B.; Earl, D.; Nguyen, N.; Diekhans, M.; Zerbino, D.; Haussler, D. Cactus: Algorithms for genome multiple sequence alignment. Genome Res. 2011, 21, 1512–1528. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Hunt, M.; Tsai, I.J. Inferring synteny between genome assemblies: A systematic evaluation [Internet]. BMC Bioinform. 2018. [CrossRef] [PubMed]
- R Core Team. R: A language and environment for statistical computing [Internet]. Vienna, Austria. 2019. Available online: https://www.R.-project.org/ (accessed on 22 August 2019).
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. circlize Implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed]
- Gel, B.; Serra, E. karyoploteR: An R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 2017, 33, 3088–3090. [Google Scholar] [CrossRef]
- Peona, V.; Weissensteiner, M.H.; Suh, A. How complete are “complete” genome assemblies?—An avian perspective. Mol. Ecol. Resour. 2018, 18, 1188–1195. [Google Scholar] [CrossRef]
- Wright, N.A.; Gregory, T.R.; Witt, C.C. Metabolic “engines” of flight drive genome size reduction in birds. Proc. Biol. Sci. 2014, 281, 20132780. [Google Scholar] [CrossRef] [PubMed]
- Dolezel, J.; Bartos, J.; Voglmayr, H.; Greilhuber, J. Nuclear DNA content and genome size of trout and human. Cytom. Part A J. Int. Soc. Anal. Cytol. 2003, 51, 127–128, author reply 129. [Google Scholar]
- Li, H. Seqtk: A fast and lightweight tool for processing FASTA or FASTQ sequences. Available online: https://github.com/lh3/seqtk (accessed on 22 August 2019).
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Zdobnov, E.M.; Kriventseva, E.V. Correlating traits of gene retention, sequence divergence, duplicability and essentiality in vertebrates, arthropods, and fungi. Genome Biol. Evol. 2011, 3, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Tegenfeldt, F.; Li, J.; Zdobnov, E.M.; Kriventseva, E.V. OrthoDB: A hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res. 2013, 41, D358–D365. [Google Scholar] [CrossRef] [PubMed]
- Suh, A.; Paus, M.; Kiefmann, M.; Churakov, G.; Franke, F.A.; Brosius, J.; Kriegs, J.O.; Schmitz, J. Mesozoic retroposons reveal parrots as the closest living relatives of passerine birds. Nat. Commun. 2011, 2, 443. [Google Scholar] [CrossRef]
- Faircloth, B.C.; McCormack, J.E.; Crawford, N.G.; Harvey, M.G.; Brumfield, R.T.; Glenn, T.C. Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Syst. Biol. 2012, 61, 717–726. [Google Scholar] [CrossRef]
- Faircloth, B.C. PHYLUCE is a software package for the analysis of conserved genomic loci. Bioinformatics 2016, 32, 786–788. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Eaton, D.A.R.; Overcast, I. ipyrad: Interactive assembly and analysis of RADseq data sets. WWW document. Available online: http://ipyrad.readthedocs.io/ (accessed on 1 August 2016).
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Andrews, C.B.; Gregory, T.R. Genome size is inversely correlated with relative brain size in parrots and cockatoos. Genome 2009, 52, 261–267. [Google Scholar] [CrossRef]
- Weissensteiner, M.H.; Pang, A.W.C.; Bunikis, I.; Höijer, I.; Vinnere-Petterson, O.; Suh, A.; Wolf, J.B.W. Combination of short-read, long-read, and optical mapping assemblies reveals large-scale tandem repeat arrays with population genetic implications. Genome Res. 2017, 27, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, E.C.; Christidis, L. The Howard and Moore complete checklist of the birds of the World: Passerines; Aves Press: Eastbourne, UK, 2014. [Google Scholar]
- O’Connor, R.E.; Farré, M.; Joseph, S.; Damas, J.; Kiazim, L.; Jennings, R.; Bennett, S.; Slack, E.A.; Allanson, E.; Larkin, D.M.; et al. Chromosome-level assembly reveals extensive rearrangement in saker falcon and budgerigar, but not ostrich, genomes. Genome Biol. 2018, 19, 171. [Google Scholar]
- Laine, V.N.; Gossmann, T.I.; van Oers, K.; Visser, M.E.; Groenen, M.A.M. Exploring the unmapped DNA and RNA reads in a songbird genome. BMC Genom. 2019, 20, 19. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Freudenberg, J. Mappability and read length. Front. Genet. 2014, 5, 381. [Google Scholar] [CrossRef]
- Prost, S.; Armstrong, E.E.; Nylander, J.; Thomas, G.W.C.; Suh, A.; Petersen, B.; Dalen, L.; Benz, B.W.; Blom, M.P.K.; Palkopoulou, E.; et al. Comparative analyses identify genomic features potentially involved in the evolution of birds-of-paradise. Gigascience 2019, 8. [Google Scholar] [CrossRef]
- Andersen, M.J.; McCullough, J.M.; Mauck, W.M.I.I.I.; Smith, B.T.; Moyle, R.G. A phylogeny of kingfishers reveals an Indomalayan origin and elevated rates of diversification on oceanic islands. J. Biogeogr. 2018, 45, 269–281. [Google Scholar] [CrossRef]
- White, N.D.; Mitter, C.; Braun, M.J. Ultraconserved elements resolve the phylogeny of potoos (Aves: Nyctibiidae). J. Avian Biol. 2017, 48, 872–880. [Google Scholar] [CrossRef]
- Moyle, R.G.; Oliveros, C.H.; Andersen, M.J.; Hosner, P.A.; Benz, B.W.; Manthey, J.D.; Travers, S.L.; Brown, R.M.; Faircloth, B.C. Tectonic collision and uplift of Wallacea triggered the global songbird radiation. Nat. Commun. 2016, 7, 12709. [Google Scholar] [CrossRef] [PubMed]
- Manthey, J.D.; Campillo, L.C.; Burns, K.J.; Moyle, R.G. Comparison of Target-Capture and Restriction-Site Associated DNA Sequencing for Phylogenomics: A Test in Cardinalid Tanagers (Aves, Genus: Piranga). Syst. Biol. 2016, 65, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Musher, L.J.; Cracraft, J. Phylogenomics and species delimitation of a complex radiation of Neotropical suboscine birds (Pachyramphus). Mol. Phylogenet. Evol. 2018, 118, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Andermann, T.; Fernandes, A.M.; Olsson, U.; Töpel, M.; Pfeil, B.; Oxelman, B.; et al. Allele Phasing Greatly Improves the Phylogenetic Utility of Ultraconserved Elements. Syst. Biol. 2019, 68, 32–46. [Google Scholar]
- Bukowicki, M.; Franssen, S.U.; Schlötterer, C. High rates of phasing errors in highly polymorphic species with low levels of linkage disequilibrium. Mol. Ecol. Resour. 2016, 16, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Cariou, M.; Duret, L.; Charlat, S. Is RAD-seq suitable for phylogenetic inference? An in silico assessment and optimization. Ecol. Evol. 2013, 3, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Eaton, D.A.R.; Spriggs, E.L.; Park, B.; Donoghue, M.J. Misconceptions on Missing Data in RAD-seq Phylogenetics with a Deep-scale Example from Flowering Plants. Syst. Biol. 2017, 66, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Del Hoyo, J.; Elliott, A.; Sargatal, J.; Christie, D.A.; de Juana, E. Handbook of the birds of the world alive; Lynx Edicions: Barcelona, Spain, 2014. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Order | Species | N50 scaf (Mb) | N50 Contig | Coverage | BioProject | Accession | Size (Gb) | Scaffolds |
|---|---|---|---|---|---|---|---|---|
| Galliformes | Gallus gallus | 90.11 | 639,813 | 248.3× | PRJNA412424 | GCA_002798355.1 | 1.02 | 1822 |
| Columbiformes | Columba livia | 24.54 | 27,697 | 60× | PRJNA347893 | GCA_001887795.1 | 1.02 | 91 |
| Apodiformes | Calypte anna | 74.1 | 14,522,327 | 54× | PRJNA489139 | GCA_003957555.1 | 1.06 | 159 |
| Falconiformes | Falco peregrinus | 26.78 | 33,994 | 137.6 | PRJNA347893 | GCA_001887755.1 | 1.11 | 72 |
| Psittaciformes | Strigops habroptila | 83.2 | 9,454,100 | 76.1 | PRJNA489135 | GCA_004027225.1 | 1.17 | 100 |
| Passeriformes | Taeniopygia guttata | 70.43 | 11,998,827 | 88.2 | PRJNA489098 | GCA_003957565.1 | 1.06 | 134 |
| Passeriformes | Ficedula albicollis | 6.54 | 410,964 | 60× | PRJNA208061 | GCA_000247815.2 | 1.12 | 21836 |
| Passeriformes | Parus major | 71.37 | 148,693 | 95× | PRJNA312399 | GCA_001522545.3 | 1.02 | 1675 |
| Passeriformes | Passer domesticus | 6.37 | 51,426 | 130× | PRJNA255814 | GCA_001700915.1 | 1.04 | 2571 |
| Passeriformes | Rhegmatorhina melanosticta | 3.3 | 136,760 | 38× | PRJNA561634 | Pending | 1.03 | 165 |
| Ragout | Chicken | Rock Pigeon | Anna’s Hummingbird | PeregrineFalcon | Kakapo * | Collard Flycatcher * | Great Tit * | House Sparrow * | Zebra Finch * | |
|---|---|---|---|---|---|---|---|---|---|---|
| Mapped | 595 | 662 | 577 | 622 | 676 | 691 | 667 | 691 | 677 | 695 |
| Unplaced | 120 | 53 | 138 | 93 | 39 | 24 | 48 | 24 | 38 | 20 |
| Chromosomes | 27 | 34 | 29 | 33 | 32 | 30 | 31 | 38 | 30 | 31 |
| Gaps inserted (Mb) | 42.8 | 0.18 | 0.12 | 0.15 | 0.16 | 0.18 | 0.16 | 0.17 | 0.16 | 0.16 |
| Taxon | Scaffolds | Expected Size (Gb) | Assembly Size (Gb) | Missing (Mb) | Gaps (Mb) | % Missing |
|---|---|---|---|---|---|---|
| Calypte anna | 159 | 1.14 | 1.06 | 80.3 | 16.1 | 8.5 |
| Corvus cornix | 145 | 1.19 | 1.05 | 144 | 9.6 | 12.9 |
| Gallus gallus | 1821 | 1.25 | 1.02 | 199 | 19.2 | 19.9 |
| Rhegmatorhina melanosticta | 715 | 1.3 | 1.03 | 270 | 12.2 | 21 |
| Strigops habroptilus | 99 | 1.28 | 1.17 | 115.5 | 27.6 | 11.2 |
| Taeniopygia guttata | 134 | 1.22 | 1.06 | 192 | 2.3 | 13.6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, L.A.; Musher, L.J.; Cracraft, J. A Multireference-Based Whole Genome Assembly for the Obligate Ant-Following Antbird, Rhegmatorhina melanosticta (Thamnophilidae). Diversity 2019, 11, 144. https://doi.org/10.3390/d11090144
Coelho LA, Musher LJ, Cracraft J. A Multireference-Based Whole Genome Assembly for the Obligate Ant-Following Antbird, Rhegmatorhina melanosticta (Thamnophilidae). Diversity. 2019; 11(9):144. https://doi.org/10.3390/d11090144
Chicago/Turabian StyleCoelho, Laís A., Lukas J. Musher, and Joel Cracraft. 2019. "A Multireference-Based Whole Genome Assembly for the Obligate Ant-Following Antbird, Rhegmatorhina melanosticta (Thamnophilidae)" Diversity 11, no. 9: 144. https://doi.org/10.3390/d11090144
APA StyleCoelho, L. A., Musher, L. J., & Cracraft, J. (2019). A Multireference-Based Whole Genome Assembly for the Obligate Ant-Following Antbird, Rhegmatorhina melanosticta (Thamnophilidae). Diversity, 11(9), 144. https://doi.org/10.3390/d11090144