Differences in Bacterial Diversity, Composition and Function due to Long-Term Agriculture in Soils in the Eastern Free State of South Africa

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
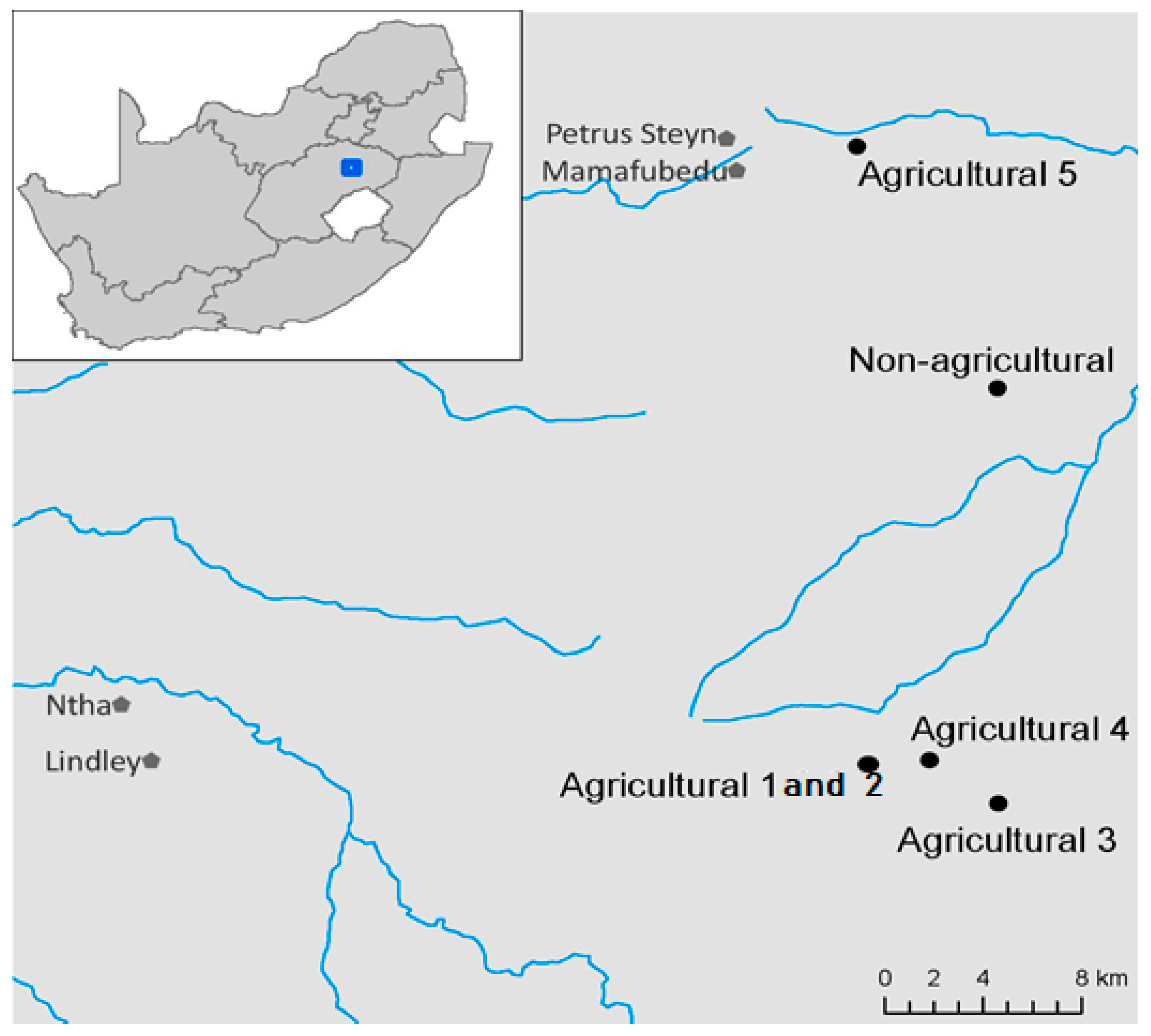
2.1. Site Description and Sampling
2.2. Soil Chemical Analyses
2.3. DNA Extraction
2.4. 16S rRNA Gene Sequencing
2.5. 16S Amplicon Analysis
2.6. Bacterial Community Analysis
2.7. Community Gene Function Prediction
3. Results
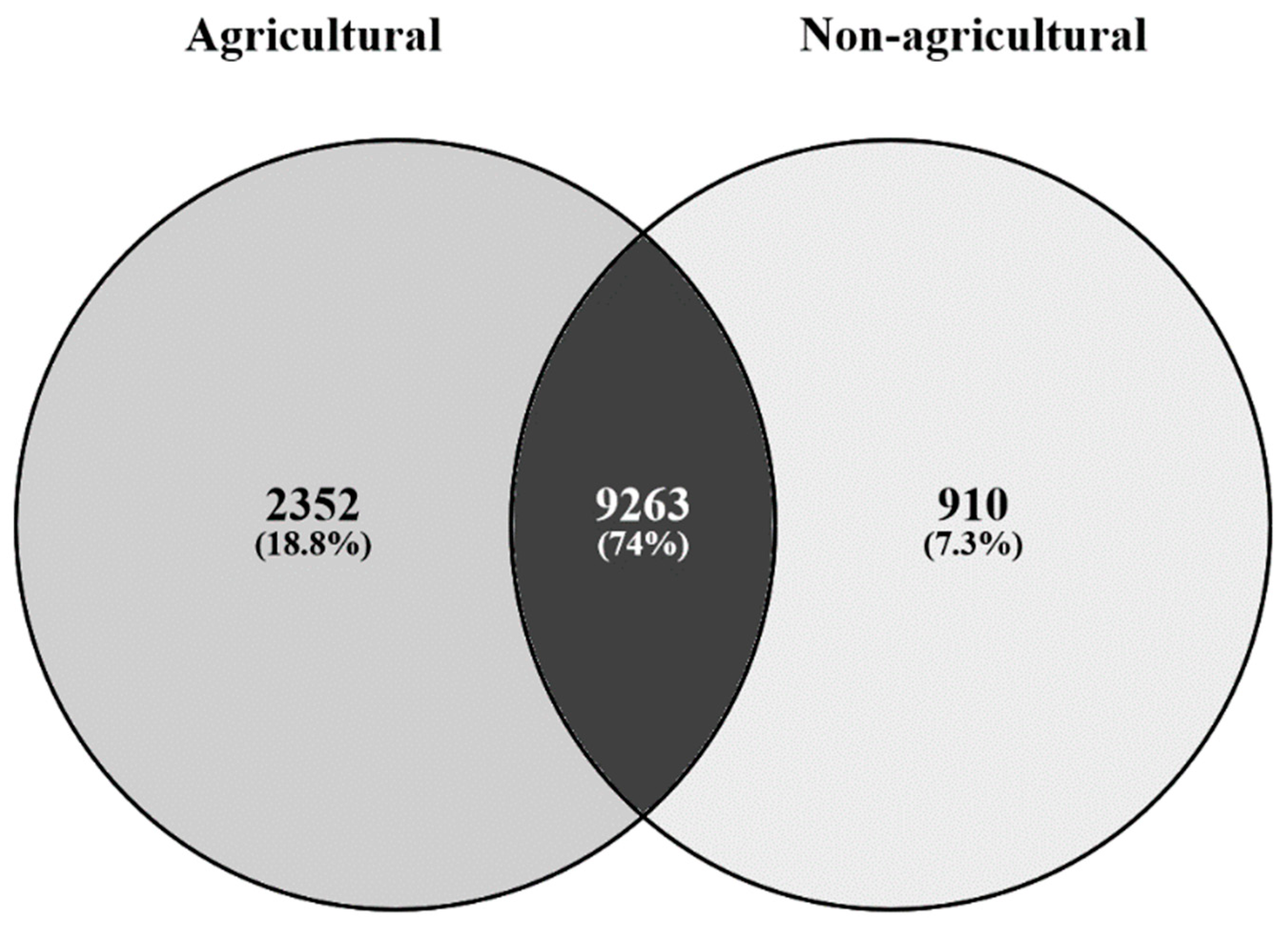
3.1. Bacterial Communities from Agricultural and Non-Agricultural Soils
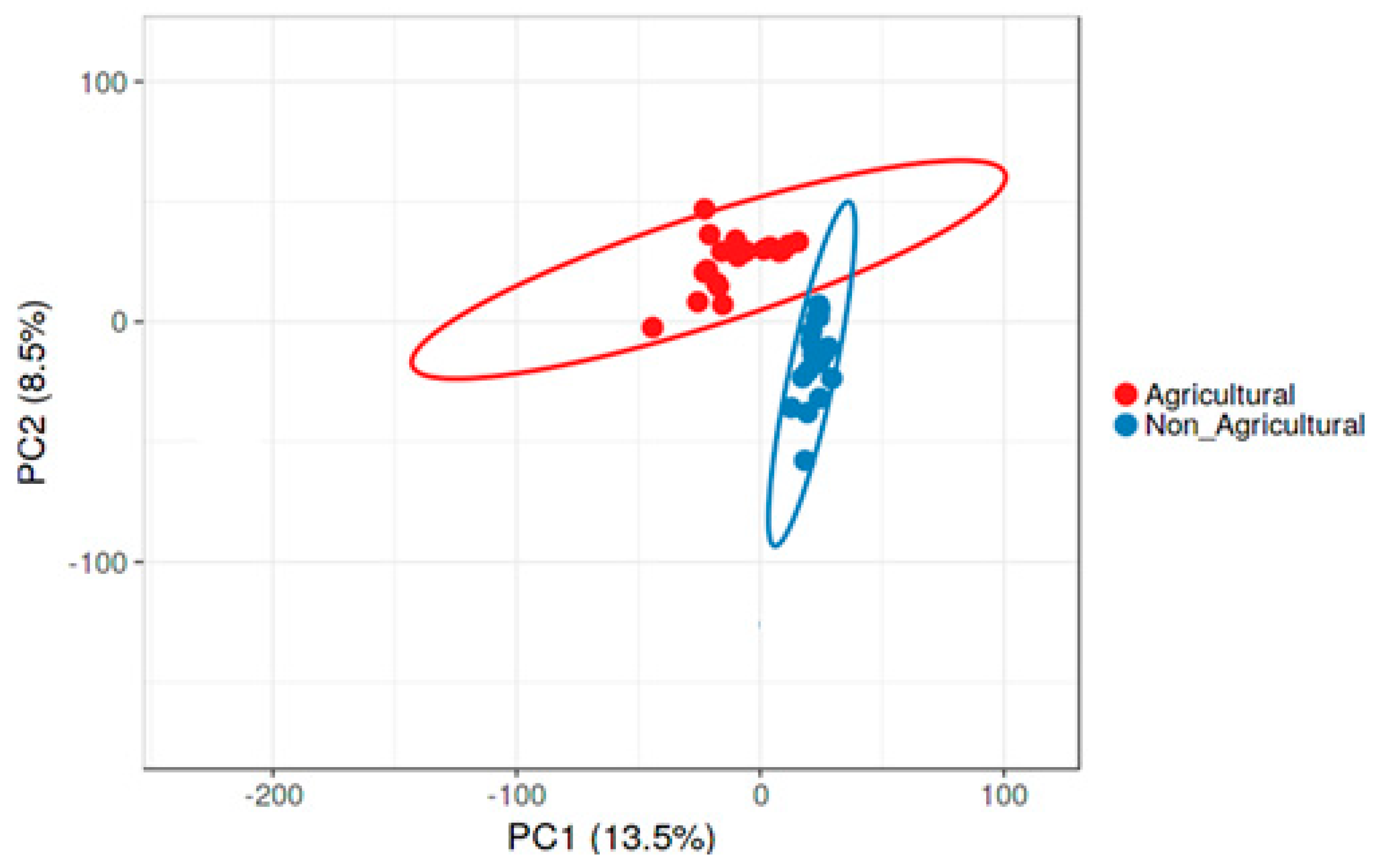
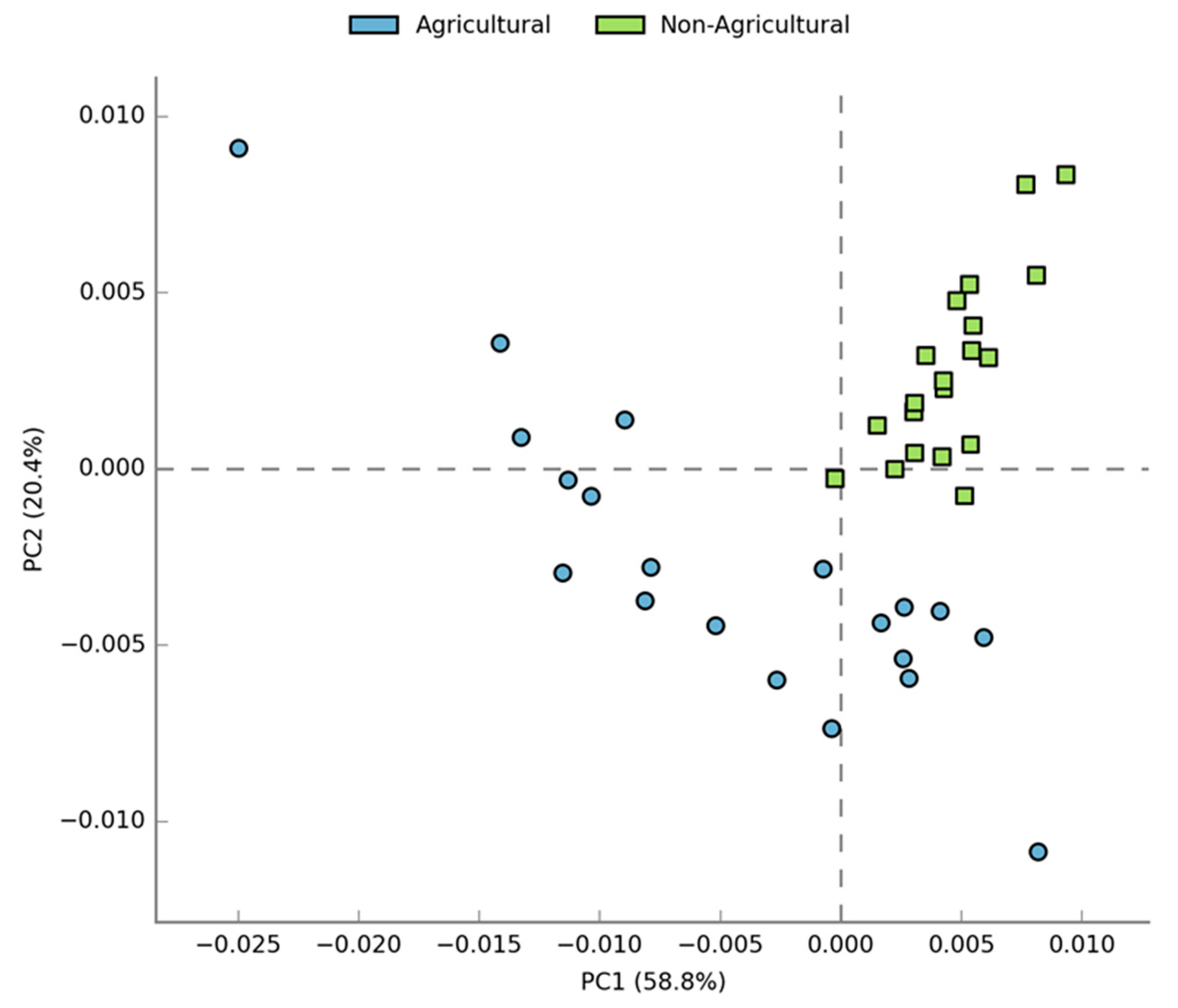
3.2. Compositional Differences in Bacterial Diversity Between Agricultural and Non-agricultural Soil
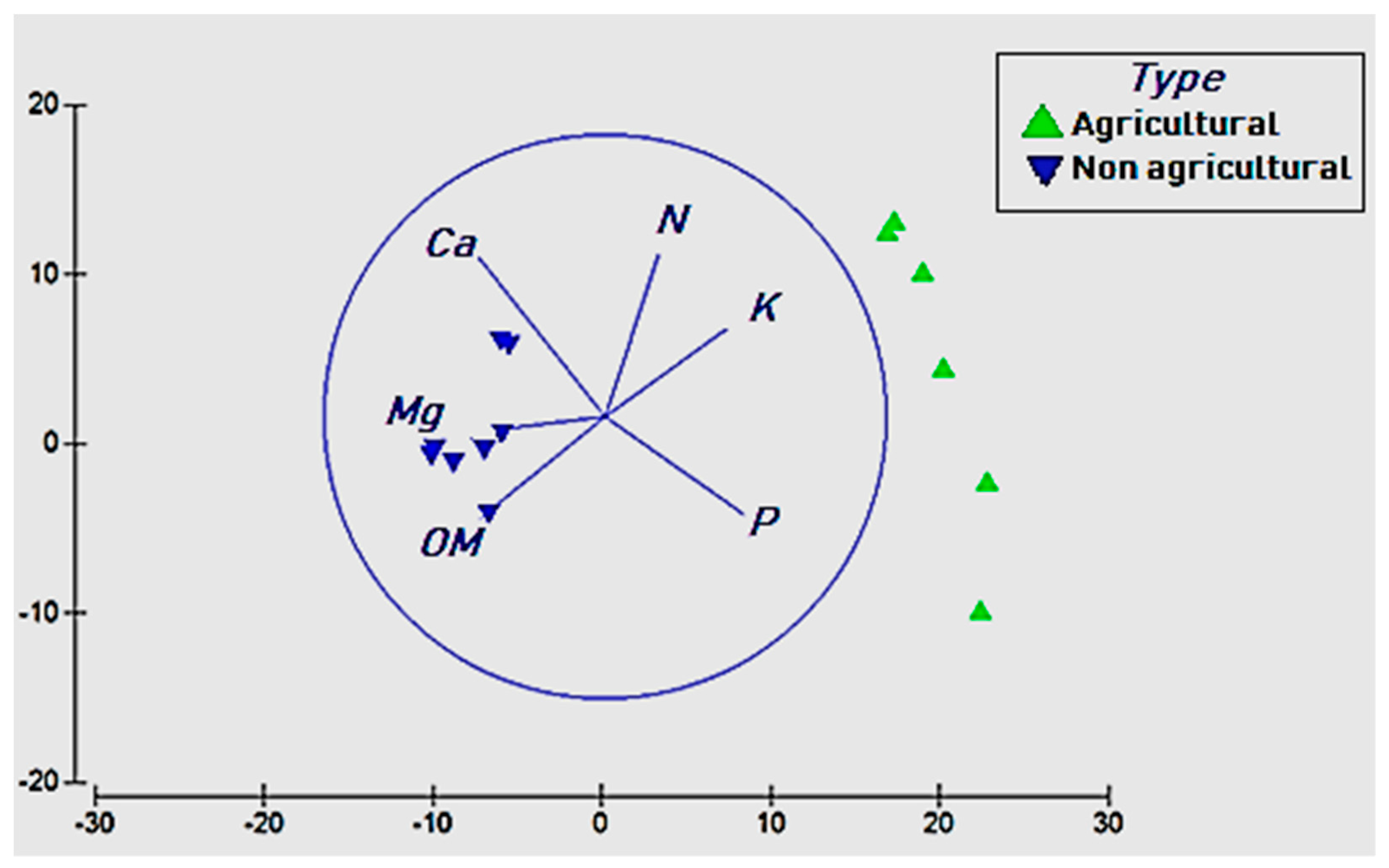
3.3. Effects of Soil Chemistry on Bacterial Diversity
3.4. Rarefaction Curves and Diversity Parameters
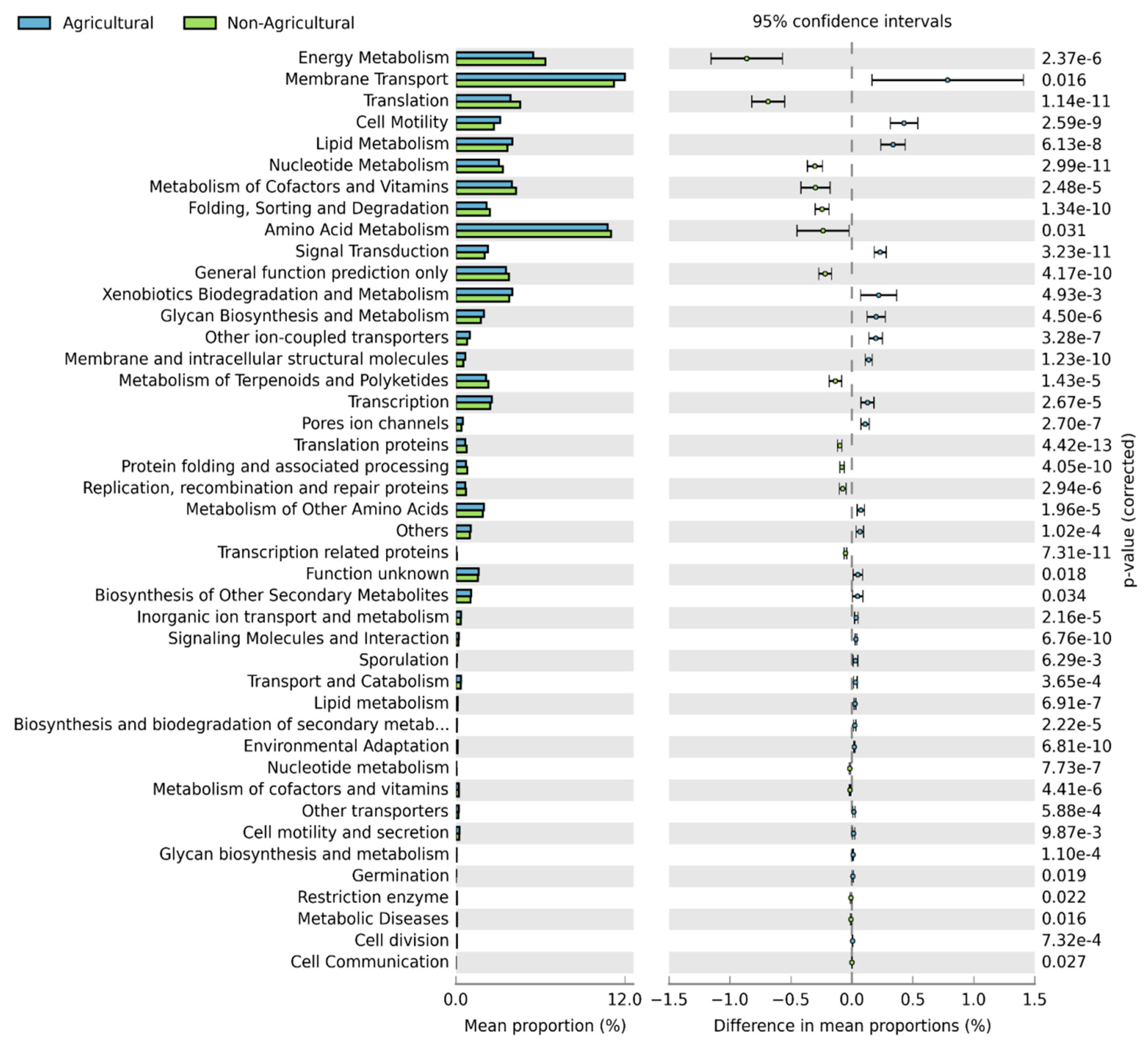
3.5. Functional Predictions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- United Nations Department of Economic and Social Affairs (UNDESA) 2017. Available online: https://www.un.org/development/desa/en/news/population/world-population-prospects-2017.html (accessed on 20 June 2018).
- Hunter, M.C.; Smith, R.G.; Schipanski, M.E.; Atwood, L.W.; Mortensen, D.A. Agriculture in 2050: Recalibrating Targets for Sustainable Intensification. BioScience 2017, 67, 386–391. [Google Scholar] [CrossRef]
- Statistics South Africa (Stats S. A.). Statistical Release P0302: Mid-Year Population Estimates 2018. 2018. Available online: http://www.statssa.gov.za/?p=11341.pdf (accessed on 24 August 2018).
- Strydom, N.; Struweg, J. Agricultural Output Versus SA’s Population Growth. 29 July 2016 Issue of Farmer’s Weekly. Available online: https://www.farmersweekly.co.za/opinion/by-invitation/agricultural-output-versus-sas-population-growth/ (accessed on 15 May 2015).
- Department of Agriculture, Forestry and Fisheries (DAFF). Abstract of Agricultural Statistics. 2018. Available online: http://www.daff.gov.za/Daffweb3/Portals/0/Statistics%20and%20Economic%20Analysis/Statistical%20Information/Abstract%202018.pdf (accessed on 27 August 2018).
- Department of Agriculture and Rural Development (DARD). State of Readiness 2017/2018. Portfolio Committee on Agriculture, Forestry and Fisheries, Capetown. 2018. Available online: http://pmg-assets.s3-website-eu-west-1.amazonaws.com/171017freestate.pdf (accessed on 27 August 2018).
- Department of Agriculture, Forestry and Fisheries (DAFF). Free State: Provincial Overview. 2015. Available online: http://www.culturalguiding.com/Courses/FreeStateProvince/ProvincialOverview/ProvincialOverview/1577.pdf (accessed on 20 June 2015).
- Maphalla, L.T.; Salman, M.V. Provincial Report on Education and Training for Agriculture and Rural Development: Free State Province. 2002. Available online: https://www.nda.agric.za/doaDev/sideMenu/educationAndTraining/Free%20State.pdf (accessed on 12 April 2018).
- Knot, J. Conservation Agriculture and Commercial Farmers in the Eastern Free State: Promoting Conservation Agriculture in the Eastern Free State of South Africa. Ph.D. Thesis, University of Free State, Bloemfontein, South Africa, 2014. [Google Scholar]
- Branca, G.; McCarthy, N.; Lipper, L.; Jolejole, M.C. Climate-Smart Agriculture: A Synthesis of Empirical Evidence of Food Security and Mitigation Benefits from Improved Cropland Management. Mitigation of Climate Change in Agriculture Series 3: p1–43. 2015. Available online: http://www.fao.org/docrep/015/i2574e/i2574e00.pdf (accessed on 18 May 2015).
- Foley, J.A.; DeFries, R.; Asner, G.P.; Barford, C.; Bonan, G.; Carpenter, S.R.; Chapin, F.S.; Coe, M.T.; Daily, G.C.; Gibbs, H.K.; et al. Global Consequences of Land-Use. Science 2005, 309, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Torsvik, V.; Daae, F.L.; Sandaa, R.A.; Ovreas, L. Novel techniques for analysing microbial diversity in natural and perturbed environments. J. Bacteriol. 1998, 64, 53–62. [Google Scholar] [CrossRef]
- Weibull, A.C.; Ostman, O.; Granqvist, A. Species richness in agroecosystems: The effect of landscape, habitat and farm management. Biodivers. Conserv. 2003, 12, 1335–1355. [Google Scholar] [CrossRef]
- Balota, E.L.; Colozzi, A.; Andrade, D.S.; Dick, R.P. Long-term tillage and crop rotation effects on microbial biomass and C and N mineralization in a Brazilian Oxisol. Soil Tillage Res. 2004, 77, 137–145. [Google Scholar] [CrossRef]
- Trivedi, P.; Delgado-Baquerizo, M.; Anderson, I.C.; Singh, B.K. Response of Soil Properties and Microbial Communities to Agriculture: Implications for Primary Productivity and Soil Health Indicators. Front. Plant Sci. 2016, 7, 990. [Google Scholar] [CrossRef]
- Osborn, A.M.; Moore, E.R.; Timmis, K.N. An evaluation of terminal-restriction fragment length polymorphism (T-RFLP) analysis for the study of microbial community structure and dynamics. Environ. Microbiol. 2000, 2, 39–50. [Google Scholar] [CrossRef]
- Rastogi, G.; Sani, R.K. Molecular Techniques to Assess Microbial Community Structure, Function and Dynamics in the Environment. In Microbes and Microbial Technology: Agricultural and Environmental Applications; Ahmad, I., Ahmad, F., Pichtel, J., Eds.; Springer Science+Business Media LLC: New York, NY, USA, 2011; pp. 29–58. [Google Scholar] [CrossRef]
- Mehlich, A. Uniformity of expressing soil test results. A case for calculating results on a volume basis. Commun. Soil Sci. Plant Anal. 1972, 3, 417–424. [Google Scholar] [CrossRef]
- Bray, R.H.; Kurtz, L.T. Determining of total, Organic and available forms of phosphorus in Soils. Soil Sci. 1945, 59, 39–45. [Google Scholar] [CrossRef]
- Al-Busaidi, A.; Yamamoto, T.; Bakheit, C.; Cookson, P. Soil Salinity Assessment by some destructive and non-destructive methods in calcareous soils. J. Jpn. Soc. Soil Phys. 2006, 104, 27–40. [Google Scholar]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 15, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.M.R.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Stevens, M.H.H.; Oksanen, M.J.; Suggests, M. The Vegan package. Community Ecol. Package 2007, 10, 631–637. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Vienna, Austria, 2013. Available online: https://www.R-project.org/ (accessed on 20 November 2017).
- Visual and Analysis of Microbial Population Structures (VAMPS). Bay Paul Center, MBL, 2016. Available online: https://vamps.mbl.edu/utils/login.php (accessed on 4 January 2017).
- De Rosario-Martinez, H. Phia: Post-Hoc Interaction Analysis. R package version 0.1–5. 2013. 2015. Available online: https://github.com/heliosdrm/phia (accessed on 21 November 2017).
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Ondov, B.; Bergman, N.; Phillippy, A. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12. [Google Scholar] [CrossRef]
- Langille, M.; Douglas, G. PICRUSt Tutorial. Available online: https://github.com/LangilleLab/microbiome_helper/wiki/PICRUSt-tutorial (accessed on 2 December 2017).
- De Voogd, N.; Cleary, D.; Polónia, A.; Gomes, N. Bacterial community composition and predicted functional ecology of sponges, sediment and seawater from the thousand islands reef complex, West Java, Indonesia. FEMS Microbiol. Ecol. 2015, 91. [Google Scholar] [CrossRef]
- Langille, M.; Zaneveld, J.; Caporaso, J.; McDonald, D.; Knights, D.; Reyes, J.; Clemente, J.; Burkepile, D.; Vega Thurber, R.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Lemanceau, P.; Blouin, M.; Muller, D.; Moënne-Loccoz, Y. Let the Core Microbiota Be Functional. Trends Plant Sci. 2017, 22, 583–595. [Google Scholar] [CrossRef] [PubMed]
- White, J.R.; Nagarajan, N.; Pop, M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 2009, 5, e1000352. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.H. Identifying the Dominant Soil Bacterial Taxa in Libraries of 16S rRNA and 16S rRNA Genes. Appl. Environ. Microbiol. 2006, 72, 1719–1728. [Google Scholar] [CrossRef]
- Delgado-Baquerizo, M.; Oliverio, A.M.; Brewer, T.E.; Benavent-Gonzalez, A.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K.; Fierer, N. A global atlas of the dominant bacteria found in soil. Science 2018, 359, 320–325. [Google Scholar] [CrossRef]
- Dai, Z.; Su, W.; Chen, H.; Barberan, A.; Zhao, H.; Yu, M.; Yu, L.; Brookes, P.C.; Schadt, C.W.; Chang, S.X.; et al. Long-term nitrogen fertilization decreases bacterial diversity and favours the growth of Actinobacteria and Proteobacteria in agro-ecosystems across the globe. Glob. Chang. Biol. 2018, 24, 3452–3461. [Google Scholar] [CrossRef]
- Lugtenberg, B.; Kamilova, F. Plant-growth-promoting rhizobacteria. Annu. Rev. Microbiol. 2009, 63, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, C.; Yu, W.; Turak, A.; Chen, D.; Huang, Y.; Ao, J.; Jiang, Y.; Huang, Z. Effects of Nitrogen and Phosphorus Inputs on Soil Bacterial Abundance, Diversity, and Community Composition in Chinese Fir Plantations. Front. Microbiol. 2018, 9, 1543. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Castro, V.H.L.; Schroeder, L.F.; Quirino, B.F.; Kruger, R.H.; Barreto, C.C. Acidobacteria from oligotrophic soil from the Cerrado can grow in a wide range of carbon source concentrations. Can. J. Microbiol. 2013, 59, 746–753. [Google Scholar] [CrossRef]
- Song, H.; Song, W.; Kim, M.; Tripathi, B.; Kim, H.; Jablonski, P.; Adams, J. Bacterial strategies along nutrient and time gradients, revealed by metagenomic analysis of laboratory microcosms. FEMS Microbiol. Ecol. 2017, 93, fix114. [Google Scholar] [CrossRef]
- Mahamane, L.; Mahamane, S. Biodiversity of ligneous species in semi-arid to arid ones of southwestern Niger according to anthropogenic and natural factors. Agric. Ecosyst. Environ. 2005, 105, 267–271. [Google Scholar] [CrossRef]
- Underwood, E.C.; Viers, J.H.; Klausmeyer, K.R.; Cox, R.L.; Shaw, M.R. Threats and biodiversity in the Mediterranean biome. Divers. Distrib. 2009, 15, 188–197. [Google Scholar] [CrossRef]
- Ding, G.C.; Piceno, Y.M.; Heuer, H.; Weinert, N.; Dohrmann, A.B.; Carrillo, A.; Andersen, G.L.; Castellanos, T.; Tebbe, C.; Smalla, K. Changes of soil bacterial diversity as a consequence of agricultural land-use in a semi-arid ecosystem. PLoS ONE 2013, 8, e59497. [Google Scholar] [CrossRef] [PubMed]
- Dudley, N.; Alexander, S. Agriculture and biodiversity: A review. Biodiversity 2017, 18, 45–49. [Google Scholar] [CrossRef]
- Altieri, M.A. The ecological role of biodiversity in agroecosystems. Agric. Ecosyst. Environ. 1999, 74, 19–31. [Google Scholar] [CrossRef]
- Szoboszlay, M.; Dohrmann, A.; Poeplau, C.; Don, A.; Tebbe, C. Impact of land-use change and soil organic carbon quality on microbial diversity in soils across Europe. FEMS Microbiol. Ecol. 2017, 93, fix146. [Google Scholar] [CrossRef] [PubMed]
- Tiemann, L.; Grandy, A.; Atkinson, E.; Marin-Spiotta, E.; McDaniel, M. Crop rotational diversity enhances belowground communities and functions in an agroecosystem. Ecol. Lett. 2015, 18, 761–771. [Google Scholar] [CrossRef]
- Carbonetto, B.; Rascovan, N.; Álvarez, R.; Mentaberry, A.; Vázquez, M.P. Structure, composition and metagenomic profile of soil microbiomes associated to agricultural land-use and tillage systems in Argentine pampas. PLoS ONE 2014, 9, e99949. [Google Scholar] [CrossRef]
- Liiri, M.; Hasab, M.; Haimi, J.; Setala, H. History of land-use intensity can modify the relationship between functional complexity of the soil fauna and soil ecosystem services—A microcosm study. Appl. Soil Ecol. 2012, 55, 53–61. [Google Scholar] [CrossRef]
- Steenwerth, K.L.; Jackson, L.E.; Caldero, F.J.; Stromberg, M.R.; Scow, K.M. Soil microbial community composition and land-use history in cultivated and grassland ecosystems of coastal California. Soil Biol. Biochem. 2002, 34, 1599–1611. [Google Scholar] [CrossRef]
- Nemergut, D.R.; Townsend, A.R.; Sattin, S.R.; Freeman, K.R.; Fierer, N.; Neff, J.C.; Bowman, W.D.; Schadt, C.W.; Weintraub, M.N.; Schmidt, S.K. The effects of chronic nitrogen fertilization on alpine tundra soil microbial communities: Implications for carbon and nitrogen cycling. Environ. Microbiol. 2008, 10, 3093–3105. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, K.S.; Lauber, C.L.; Knight, R.; Bradford, M.A.; Fierer, N. Consistent effects of nitrogen fertilization on soil bacterial communities in contrasting systems. Ecology 2010, 91, 3463–3470. [Google Scholar] [CrossRef]
- Fierer, N.; Leff, J.W.; Adams, B.J.; Nielsen, U.N.; Bates, S.T.; Lauber, C.L.; Owens, S.; Jack, G.A.; Wall, D.H.; Caporaso, J.G. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. USA 2012, 109, 21390–21395. [Google Scholar] [CrossRef] [PubMed]
- García-Orenes, F.; Morugán-Coronado, A.; Zornoza, R.; Scow, K. Changes in Soil Microbial Community Structure Influenced by Agricultural Management Practices in a Mediterranean Agro-Ecosystem. PLoS ONE 2013, 8, e80522. [Google Scholar] [CrossRef] [PubMed]
- Knelman, J.E.; Schmidt, S.K.; Lynch, R.C.; Darcy, J.L.; Castle, S.C.; Cleveland, C.C.; Nemergut, D.R. Nutrient addition dramatically accelerates microbial community succession. PLoS ONE 2014, 9, e102609. [Google Scholar] [CrossRef] [PubMed]
- Ladd, J.N.; Foster, R.C.; Skjemstad, J.O. Soil structure: Carbon and nitrogen metabolism. Geoderma 1993, 56, 401–434. [Google Scholar] [CrossRef]
- Beare, M.H.; Cabrera, M.L.; Hendrix, P.F.; Coleman, D.C. Aggregate-protected and unprotected organic matter pools in conventional and no-tillage soil. Soil Sci. Soc. Am. J. 1994, 58, 787–795. [Google Scholar] [CrossRef]
- Bardgett, R.D.; McAlister, E. The measurement of soil fungal: Bacterial biomass ratios as an indicator of ecosystem self-regulation in temperate meadow grasslands. Biol. Fertil. Soils 1999, 29, 282–290. [Google Scholar] [CrossRef]
- Bot, A.; Benites, J. The Importance of Soil Organic Matter; FAO: Viale delle Terme di Caracalla, Rome, Italy, 2005; ISBN 9251053669. [Google Scholar]
- Lal, R. Soil carbon stocks under present and future climate with specific reference to European ecoregions. Nutr. Cycl. Agroecosyst. 2008, 81, 113–127. [Google Scholar] [CrossRef]
- Smith, P. Land-use change and soil organic carbon dynamics. Nutr. Cycl. Agroecosyst. 2008, 81, 169–178. [Google Scholar] [CrossRef]
- Mishra, U.; Ussiri, D.A.N.; Lal, R. Tillage effects on soil organic carbon storage and dynamics in Corn Belt of Ohio, USA. Soil Tillage Res. 2010, 107, 88–96. [Google Scholar] [CrossRef]
- Glanz, J. Saving Our Soil: Solutions for Sustaining Earth’s Vital Resource; Johnson Printing: Boulder, CO, USA, 1995. [Google Scholar]
- Taghipour, A.; Rezapour, S.; Dovlati, B.; Hamzenejad, R. Effects of Land-use Changes on Some Soil Chemical Properties in Khoy, West Azerbaijan Province. Majallah-i āb va Khāk 2016, 29, 418–431. [Google Scholar] [CrossRef]
- Butterfield, C.N.; Li, Z.; Andeer, P.F.; Spaulding, S.; Thomas, B.C.; Singh, A.; Hettich, R.L.; Suttle, K.B.; Probst, A.J.; Tringe, S.G.; et al. Proteogenomic analyses indicate bacterial methylotrophy and archaeal heterotrophy are prevalent below the grass root zone. PeerJ 2016, 4, e2687. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, G.; Heuer, V.; Lazar, C.; Goldhammer, T.; Wendt, J.; SaSongmarkin, V.; Elvert, M.; Teske, A.; Joye, S.; Hinrichs, K. Relative importance of methylotrophic methanogenesis in sediments of the Western Mediterranean Sea. Geochim. Et Cosmochim. Acta 2018, 224, 171–186. [Google Scholar] [CrossRef]
- Tedla, A.B. Direct and Residual Effects of Organic and Inorganic Fertilizers on Soil Chemical Properties, Microbial Components and Maize Yield Under Long-term Crop Rotation. Ph.D. Thesis, University of Pretoria, Pretoria, South Africa, 2013. [Google Scholar]
- Staley, C.; Gould, T.; Wang, P.; Phillips, J.; Cotner, J.; Sadowsky, M. Core functional traits of bacterial communities in the Upper Mississippi River show limited variation in response to land cover. Front. Microbiol. 2014, 5, 414. [Google Scholar] [CrossRef]
- Hariharan, J.; Sengupta, A.; Grewal, P.; Dick, W. Functional Predictions of Microbial Communities in Soil as Affected by Long-term Tillage Practices. Agric. Environ. Lett. 2017, 2, 1–5. [Google Scholar] [CrossRef]
- Jansson, J.; Hofmockel, K. The soil microbiome—From metagenomics to metaphenomics. Curr. Opin. Microbiol. 2018, 43, 162–168. [Google Scholar] [CrossRef]
- Lemanceau, P.; Bakker, P.; De Kogel, W.J.; Alabouvette, C.; Schippers, B. Effect of pseudobactin 358 production by Pseudomonas putida WCS358 on suppression of Fusarium wilt of carnations by non-pathogenic Fusarium oxysporum Fo47. Appl. Environ. Microbiol. 1992, 58, 2978–2982. [Google Scholar]
- Zaccardelli, M.; De Nicola, F.; Villecco, D.; Scotti, R. The development and suppressive activity of soil microbial communities under compost amendment. J. Soil Sci. Plant Nutr. 2013, 13, 730–742. [Google Scholar] [CrossRef]
- Van Loon, L.C.; Bakker, P.A.H.M.; Pieterse, C.M.J. Systemic resistance induced by rhizosphere bacteria. Annu. Rev. Phytopathol. 1998, 36, 453–483. [Google Scholar] [CrossRef]
- Baltz, R.H. Antimicrobials from Actinomycetes: Back to the Future. Microbe 2007, 2, 125–131. Available online: https://www.semanticscholar.org/paper/Antimicrobials-from-Actinomycetes-%3A-Back-to-the-are-Baltz/b80edf2f02e88eb7cf024fa1a5c593721cb713be#citing-papers (accessed on 13 September 2018).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phylum | Agricultural | Non-Agricultural | p-Value |
|---|---|---|---|
| Proteobacteria | 40% | 34% | 3.788 × 10−5 * |
| Actinobacteria | 23% | 40% | 0.0323 * |
| Bacteroidetes | 12% | 4% | 0.0410 * |
| Chloroflexi | 8% | 6% | 0.8106 |
| Firmicutes | 7% | 4% | 0.4156 |
| Acidobacteria | 3% | 6% | 3.877 × 10−5 * |
| Diversity Index | Agricultural | Non-Agricultural |
|---|---|---|
| Richness | 598 (2.85) * | 504 (2.56) |
| ACE | 46.4 (2.78) * | 34.1 (3.20) |
| Chao1 | 44.5 (4.96) * | 33.0 (4.09) |
| Shannon | 2.80 (0.31) * | 2.58 (0.12) |
| Simpson | 0.76 (0.10) | 0.80 (0.03) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dube, J.P.; Valverde, A.; Steyn, J.M.; Cowan, D.A.; van der Waals, J.E. Differences in Bacterial Diversity, Composition and Function due to Long-Term Agriculture in Soils in the Eastern Free State of South Africa. Diversity 2019, 11, 61. https://doi.org/10.3390/d11040061
Dube JP, Valverde A, Steyn JM, Cowan DA, van der Waals JE. Differences in Bacterial Diversity, Composition and Function due to Long-Term Agriculture in Soils in the Eastern Free State of South Africa. Diversity. 2019; 11(4):61. https://doi.org/10.3390/d11040061
Chicago/Turabian StyleDube, Joel P., Angel Valverde, Joachim M. Steyn, Don A. Cowan, and Jacqueline E. van der Waals. 2019. "Differences in Bacterial Diversity, Composition and Function due to Long-Term Agriculture in Soils in the Eastern Free State of South Africa" Diversity 11, no. 4: 61. https://doi.org/10.3390/d11040061
APA StyleDube, J. P., Valverde, A., Steyn, J. M., Cowan, D. A., & van der Waals, J. E. (2019). Differences in Bacterial Diversity, Composition and Function due to Long-Term Agriculture in Soils in the Eastern Free State of South Africa. Diversity, 11(4), 61. https://doi.org/10.3390/d11040061