No Signs of Genetic Erosion in a 19th Century Genome of the Extinct Paradise Parrot (Psephotellus pulcherrimus)

 , , and
, , and
Abstract

1. Introduction
2. Materials and Methods
2.1. Extraction, Library Preparation and Sample Information
2.2. Filtering of Raw Reads and Mapping
2.3. Phylogenetic Analyses and Divergence Time Estimation
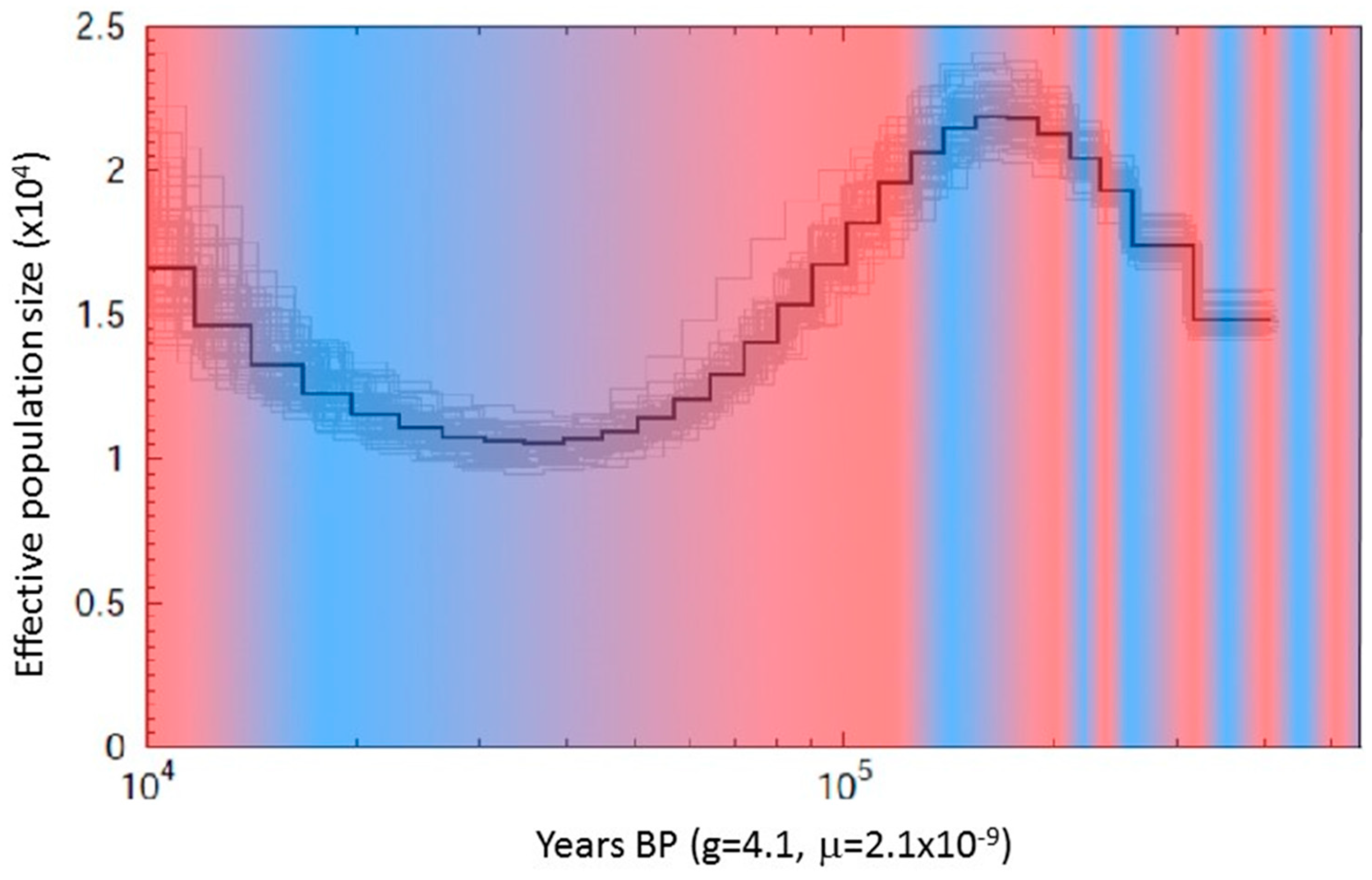
2.4. Population Estimates
3. Results
3.1. Mapped Genomes and Population Genetics
3.2. Phylogenetic Relationships and Divergence Estimates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gould, J. On a new species of Platycerus. Ann. Mag. Nat. Hist. 1845, 15, 114–115. [Google Scholar] [CrossRef][Green Version]
- Gould, J. The Birds of Australia; University of Glasgow: Scotland, UK, 1846; Available online: http://special.lib.gla.ac.uk/exhibns/month/july2005.html (accessed on 12 April 2019).
- Olsen, P.; National Library of Australia. Glimpses of Paradise: The Quest for the Beautiful Parrakeet; National Library of Australia: Canberra, Australia, 2007. [Google Scholar]
- Macdonald, J.D. Birds of Australia: A Summary of Information; HF & G. Witherby: Sydney, Australia, 1973. [Google Scholar]
- del Hoyo, J.; Elliott, A.; Sargatal, J. Handbook of the Birds of the World. Vol. 4. Sandgrouse to Cuckoos; Lynx Edicions: Barcelona, Spain, 1997. [Google Scholar]
- Forshaw, J.M.; Cooper, W.T. Australian Parrots, 3rd ed.; Alexander Editions: Robina, Australia, 2002. [Google Scholar]
- Olsen, P. The Paradise Parrot. Available online: http://www.nla.gov.au/pub/paradiseparrot/ (accessed on 12 April 2019).
- Forshaw, J.M.; Shephard, M. Grassfinches in Australia. Illustrated by Anthony Pridham; CSIRO Publishing: Melbourne, Australia, 2012. [Google Scholar]
- Garnett, S.T.; Szabo, J.K.; Dutson, G. The Action Plan for Australian Birds; CSIRO Publishing: Melbourne, Australia, 2011. [Google Scholar]
- Keller, L.F.; Waller, D.M. Inbreeding effects in wild populations. Trends Ecol. Evol. 2002, 17, 230–241. [Google Scholar] [CrossRef]
- Lande, R.; Shannon, S. The role of genetic variation in adaptation and population persistence in a changing environment. Evolution 1996, 50, 434–437. [Google Scholar] [CrossRef]
- Smith, K.F.; Sax, D.F.; Lafferty, K.D. Evidence for the role of infectious disease in species extinction and endangerment. Conserv. Biol. 2006, 20, 1349–1357. [Google Scholar] [CrossRef]
- Reed, D.H.; Frankham, R. Correlation between fitness and genetic diversity. Conserv. Biol. 2003, 17, 230–237. [Google Scholar] [CrossRef]
- van der Valk, T.; Diez-del-Molino, D.; Marques-Bonet, T.; Guschanski, K.; Dalen, L. Historical Genomes Reveal the Genomic Consequences of Recent Population Decline in Eastern Gorillas. Curr. Biol. 2019, 29, 165–170. [Google Scholar] [CrossRef]
- Bi, K.; Linderoth, T.; Vanderpool, D.; Good, J.M.; Nielsen, R.; Moritz, C. Unlocking the vault: Next-generation museum population genomics. Mol. Ecol. 2013, 22, 6018–6032. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.H.; Fang, Q.; Barnett, R.; Li, C.; Han, S.J.; Kuhlwilm, M.; Zhou, L.; Pan, H.L.; Deng, Y.; Chen, G.J.; et al. The Genomic Footprints of the Fall and Recovery of the Crested Ibis. Curr. Biol. 2019, 29, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Irestedt, M.; Ohlson, J.I.; Zuccon, D.; Kallersjö, M.; Ericson, P.G.P. Nuclear DNA from old collections of avian study skins reveals the evolutionary history of the Old World suboscines (Aves, Passeriformes). Zool. Scr. 2006, 35, 567–580. [Google Scholar] [CrossRef]
- Meyer, M.; Kircher, M. Illumina Sequencing Library Preparation for Highly Multiplexed Target Capture and Sequencing. Cold Spring Harb. Protoc. 2010. [Google Scholar] [CrossRef]
- Blom, M.P.K. GitHub. Available online: https://github.com/mozesblom (accessed on 12 April 2019).
- Zhang, J.J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef]
- Petersen, K.R.; Street, D.A.; Gerritsen, A.T.; Hunter, S.S.; Settles, M.L. Super deduper, fast PCR duplicate detection in fastq files. In Proceedings of the 6th ACM Conference on Bioinformatics, Computational Biology and Health Informatics, Atlanta, GA, USA, 9–12 September 2015; pp. 491–492. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Andrews, S. Fastqc: A Quality Control Tool for High throughput Sequence Data; Babraham Bioinformatics Group: Cambridge, UK, 2010; Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 12 April 2019).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- van der Zwan, H.; van der Westhuizen, F.; Visser, C.; van der Sluis, R. Draft De Novo Genome Sequence of Agapornis roseicollis for Application in Avian Breeding. Anim. Biotechnol. 2018, 29, 241–246. [Google Scholar] [CrossRef]
- Anmarkrud, J.A.; Lifjeld, J.T. Complete mitochondrial genomes of eleven extinct or possibly extinct bird species. Mol. Ecol. Resour. 2017, 17, 334–341. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Proc, G.P.D. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Bcftools. Available online: https://samtools.github.io/bcftools (accessed on 12 April 2019).
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- HMMER: biosequence analysis using profile hidden Markov models. Available online: http://hmmer.org (accessed on 12 April 2019).
- Schweizer, M.; Guntert, M.; Hertwig, S.T. Out of the Bassian province: Historical biogeography of the Australasian platycercine parrots (Aves, Psittaciformes). Zool. Scr. 2013, 42, 13–27. [Google Scholar] [CrossRef]
- Joseph, L.; Toon, A.; Schirtzinger, E.E.; Wright, T.F. Molecular systematics of two enigmatic genera Psittacella and Pezoporus illuminate the ecological radiation of Australo-Papuan parrots (Aves: Psittaciformes). Mol. Phylogenet. Evol. 2011, 59, 675–684. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.A.; Yu, L.L.; Edwards, S.V. A maximum pseudo-likelihood approach for estimating species trees under the coalescent model. BMC Evol. Biol. 2010, 10, 302. [Google Scholar] [CrossRef] [PubMed]
- Weir, J.T.; Schluter, D. Calibrating the avian molecular clock. Mol. Ecol. 2008, 17, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 2011, 475, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Nadachowska-Brzyska, K.; Burri, R.; Smeds, L.; Ellegren, H. PSMC analysis of effective population sizes in molecular ecology and its application to black-and-white Ficedula flycatchers. Mol. Ecol. 2016, 25, 1058–1072. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Zhang, G.J.; Li, C.; Li, Q.Y.; Li, B.; Larkin, D.M.; Lee, C.; Storz, J.F.; Antunes, A.; Greenwold, M.J.; Meredith, R.W.; et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 2014, 346, 1311–1320. [Google Scholar] [CrossRef]
- Ganapathy, G.; Howard, J.T.; Ward, J.M.; Li, J.W.; Li, B.; Li, Y.R.; Xiong, Y.Q.; Zhang, Y.; Zhou, S.G.; Schwartz, D.C.; et al. High-coverage sequencing and annotated assemblies of the budgerigar genome. Gigascience 2014, 3, 11. [Google Scholar] [CrossRef]
- Hansen, J.; Sato, M.; Russell, G.; Kharecha, P. Climate sensitivity, sea level and atmospheric carbon dioxide. Philos. Trans. R. Soc. A 2013, 371, 20120294. [Google Scholar] [CrossRef] [PubMed]
- Nadachowska-Brzyska, K.; Li, C.; Smeds, L.; Zhang, G.J.; Ellegren, H. Temporal Dynamics of Avian Populations during Pleistocene Revealed by Whole-Genome Sequences. Curr. Biol. 2015, 25, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.J.; Harle, K.J.; Gale, S.J.; Heijnis, H. The vegetation history of the last glacial-interglacial cycle in eastern New South Wales, Australia. J. Quat. Sci. 2006, 21, 735–750. [Google Scholar] [CrossRef]
- Li, S.B.; Li, B.; Cheng, C.; Xiong, Z.J.; Liu, Q.B.; Lai, J.H.; Carey, H.V.; Zhang, Q.; Zheng, H.B.; Wei, S.G.; et al. Genomic signatures of near-extinction and rebirth of the crested ibis and other endangered bird species. Genome Biol. 2014, 15, 557. [Google Scholar] [CrossRef] [PubMed]
- Franklin, D.C.; Whitehead, P.J.; Pardon, G.; Matthews, J.; McMahon, P.; McIntyre, D. Geographic patterns and correlates of the decline of granivorous birds in northern Australia. Wildl. Res. 2005, 32, 399–408. [Google Scholar] [CrossRef]
- Christidis, L.; Norman, J. Genetic Variability in the Golden-Shouldered Parrot, Psephotus Chrysopterygius; Museum of Victoria: Abbotsford, Australia, 1996. [Google Scholar]
- Groen, H.D. Australian Parakeets; Their Maintenance and Breeding in Captivity; Published by the author; Haren, The Netherlands, 1966. [Google Scholar]
- De Ross, L. Keeping Parrots in Australian Aviaries; Lansdowne: Melbourne, Australia, 1975. [Google Scholar]
- Johnson, N.K.; Cicero, C. New mitochondrial DNA data affirm the importance of Pleistocene speciation in North American birds. Evolution 2004, 58, 1122–1130. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Species | Rag1 | ZENK | c-mos | cytb | ND2 |
|---|---|---|---|---|---|
| Psephotellus chrysopterygius | JF807991 | JF807977 | JF807963 | JX442436 | JX442400 |
| Psephotellus dissimilis | GQ505210 | GQ505155 | GQ505101 | JX442415 | JX442384 |
| Psephotus haematonotus | JX442403 | JX442379 | JX442404 | JX442435 | JX442399 |
| Psephotellus varius | GQ505209 | GQ505154 | GQ505100 | JX442414 | JX442383 |
| Purpureicephalus spurius | GQ505204 | GQ505150 | GQ505096 | JX442410 | JX442381 |
| Platycercus venustus | GQ505207 | GQ505153 | GQ505098 | JX442413 | JX442382 |
| Lathamus discolor | GQ505211 | GQ505156 | GQ505102 | JX442416 | JX442385 |
| Species | SNP rate (10−3) | Ref |
|---|---|---|
| Kea, Nestor notabilis | 0.91 | [42] |
| Budgerigar, Melopsittacus undulatus | 4.31 | [43] |
| Paradise parrot, Psephotellus pulcherrimus | 1.70 | This study |
| JX442416 | JX442435 | JX442413 | JX442410 | JX442414 | JX442415 | JX442436 | |
|---|---|---|---|---|---|---|---|
| Lathamus discolor JX442416 | |||||||
| Psephotus haematonotus JX442435 | 12.67% | ||||||
| Platycercus venustus JX442413 | 11.99% | 10.16% | |||||
| Purpureicephalus spurius JX442410 | 12.10% | 10.96% | 10.05% | ||||
| Psephotellus varius JX442414 | 10.62% | 9.93% | 8.11% | 8.45% | |||
| Psephotellus dissimilis JX442415 | 12.10% | 10.39% | 9.36% | 9.59% | 7.99% | ||
| Psephotellus chrysopterygius JX442436 | 11.53% | 11.19% | 9.93% | 9.02% | 7.99% | 6.85% | |
| Psephotellus pulcherrimus this study | 11.31% | 11.31% | 11.09% | 9.76% | 7.76% | 6.21% | 1.77% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irestedt, M.; Ericson, P.G.P.; Johansson, U.S.; Oliver, P.; Joseph, L.; Blom, M.P.K. No Signs of Genetic Erosion in a 19th Century Genome of the Extinct Paradise Parrot (Psephotellus pulcherrimus). Diversity 2019, 11, 58. https://doi.org/10.3390/d11040058
Irestedt M, Ericson PGP, Johansson US, Oliver P, Joseph L, Blom MPK. No Signs of Genetic Erosion in a 19th Century Genome of the Extinct Paradise Parrot (Psephotellus pulcherrimus). Diversity. 2019; 11(4):58. https://doi.org/10.3390/d11040058
Chicago/Turabian StyleIrestedt, Martin, Per G. P. Ericson, Ulf S. Johansson, Paul Oliver, Leo Joseph, and Mozes P. K. Blom. 2019. "No Signs of Genetic Erosion in a 19th Century Genome of the Extinct Paradise Parrot (Psephotellus pulcherrimus)" Diversity 11, no. 4: 58. https://doi.org/10.3390/d11040058
APA StyleIrestedt, M., Ericson, P. G. P., Johansson, U. S., Oliver, P., Joseph, L., & Blom, M. P. K. (2019). No Signs of Genetic Erosion in a 19th Century Genome of the Extinct Paradise Parrot (Psephotellus pulcherrimus). Diversity, 11(4), 58. https://doi.org/10.3390/d11040058