Abstract
Revealing the genetic population structure in abundant avian species is crucial for understanding speciation, conservation, and evolutionary history. The Red-backed Shrike Lanius collurio, an iconic songbird renowned for impaling its prey, is widely distributed as a breeder across much of Europe, Asia Minor and western Asia. However, in recent decades, many populations have declined significantly, as a result of habitat loss, hunting along migration routes, decrease of arthropod food, and climate change e.g., severe droughts in Africa. Within this context, gene flow among different breeding populations becomes critical to ensure the survival of the species, but we still lack an overview on the genetic diversity of the species. In this paper, we analyzed the mitochondrial cytochrome b gene (mtDNA) and the cytochrome c oxidase subunit 1 gene (mtDNA) of 132 breeding Red-backed Shrikes from across the entire breeding range to address this knowledge gap. Our results revealed consistent genetic diversity and 76 haplotypes among the Eurasian populations. Birds are clustered in two major groups, with no clear geographical separation, as a direct consequence of Pleistocene glaciations and apparent lineage mixing in refugia. This has led to genetic panmixia.
1. Introduction
Quaternary cold periods have greatly shaped the evolution and distribution of present biota worldwide [1], and this is particularly evident in the northern temperate regions [2]. During the Pleistocene era—starting 2.5 million years ago and ending just 11,000 years before present—the climate in northern temperate regions was characterized by dramatic oscillations, commonly known as “glaciations” [3,4], each of them lasting anywhere between 21,000 to 100,000 years [5]. These severe glacial episodes forced biota to retreat to southern refugia [6,7], which implied admixing of populations. Subsequently, after the retreat of the ice cap, populations expanded and recolonized the northern latitudes [8,9]. This complex process of post-glacial population expansion northward from the southern refugia occurred at least 30 times during the Pleistocene [3] and was in strong interdependence with the vegetation availability [10]. As the flora and fauna recolonized northern habitats, populations from different refugia came in contact and merged, sometimes creating the so-called hybrid or suture zones [8,11]. Based on morphological features alone, the admixture of population lineages and suture zones were evident in numerous species across the Palearctic [12]. But it was only after the emergence of molecular techniques and their wide application in zoology that the history of post-glaciation colonization and its impact on the genetic structure of European biota really unfolded [13].
Across the animal kingdom, birds have a particular significance for phylogeographic and phylogenetic studies due to their high locomotive capacities, which stimulates both gene flow and speciation events but also, in many taxa, fidelity to the natal site, which slows down population admixture [14]. Furthermore, due to their ubiquitous status and charismatic nature, birds have been at the forefront of genetic studies in zoology in recent decades [15].
The Shrike family (Aves, Laniidae) is currently comprised of 34 species, belonging to four genera: Corvinella, Urosteles, Eurocephalus, and Lanius [16,17]. Several species, notably the ones from western Palearctic, are well studied, partly due to their charisma and habit of impaling their prey. Nonetheless, within the true shrikes—genus Lanius, only few taxa have been the focus of genetic studies: the Lesser Grey Shrike (L. minor) [18], the Great Grey Shrike (L. excubitor) [19], the Southern Grey Shrike (L. meridionalis) [20,21], and the Loggerhead Shrike (L. ludovicianus) from North America [22,23]. The genetic structure of the Red-backed Shrike (L. collurio), which, despite its population decline in recent decades [24] still remains the most common shrike species across Europe [25,26], has not been yet elucidated. Furthermore, the Red-backed Shrike (hereafter RBS) is illegally trapped in the Mediterranean basin in great numbers, with as many as 30,000 birds/year in the Sinai Peninsula alone [27,28]. As such, gene flow becomes important in ensuring the wellbeing of the species [29]. To approach this knowledge gap, we analyzed the mitochondrial DNA of 132 breeding RBSs from 14 countries across the breeding range. Mitochondrial DNA (hereafter mtDNA) is an efficient marker for indicating the rate of evolution and speciation, haplotype diversity, and natal origin [30,31]. For our study, we selected the cytochrome oxidase B (hereafter Cytb) and cytochrome c oxidase subunit 1 (hereafter CO1) mitochondrial genes, regarded as two of the most efficient molecular markers for avian population genetics based on mtDNA [32]. However, it should be noted that mtDNA is only maternally inherited and non-recombinant, and therefore reflects solely the evolutionary history of females. Moreover, a number of studies [33,34,35] have challenged the advantages of mtDNA usage in phylogeny, but we argue that our results, albeit derived from just 1.551 base pairs, are in agreement with similar European studies on avian phylogeography (see Discussion for more details). The objectives of our research were (i) to evaluate the genetic diversity and population structure, in conjunction with (ii) phylogeography and evolutionary history of the RBS breeding in the western Palearctic. Based on our findings, we discuss the factors that shaped the current genetic structure of RBS across its breeding range and future implications for its conservation.
2. Materials and Methods
2.1. Sampling
We collected blood (30 µL) from breeding birds (n = 17) in Germany (Rhein-Neckar area) by puncturing the brachial vein, during the 2016 to 2017 breeding seasons. We further received blood, feathers, tissue, or buccal swabs from RBSs breeding in the following countries: Belgium, Bulgaria, Czech Republic, France, Hungary, Latvia, The Netherlands, Poland, Romania, Russia, Spain, Sweden, and Ukraine. For our collaborators with no previous experience in collecting buccal swabs, we provided a video protocol on how to sample buccal mucosa from live birds (see Video S1). All samples obtained from live birds were collected by accredited bird ringers, following national regulations. Blood and buccal swabs were conserved in EDTA buffer (0.1 M Tris, pH 7.4, 10% EDTA, 1% NaF, 0.1% Thymol) or in ethanol (70%). Feathers were stored in dry envelopes. Samples from Sweden were received from the Swedish National History Museum. Samples from Russia (except 91037 to 91048) and Ukraine were obtained from the Zoological Museum of Moscow University. For each sample we allocated an internal voucher at the collection of the Institute for Pharmacy and Molecular Biotechnology (IPMB). Full details of all samples included in our study are found in Table S1.
2.2. DNA Extraction, Polymerase Chain Reaction, and Sequencing
From each sample we extracted genomic DNA following a standard phenol-chloroform protocol [36]. We used polymerase chain reaction (hereafter PCR) to amplify the mitochondrial gene Cytb and the CO1 gene. For the Cytb reaction, we used the forward primer L14764 (5ˊ TGR TAC AAA AAA ATA GGM CCM GAA GG 3ˊ) [37] and reverse primer Mt-FSH (5ˊ TAG TTG GCC AAT GAT GAT GAA TGG GTC TTC TAC TGG TT 3ˊ) [32]. For the CO1 reaction, we used the forward primer L6615 (5ˊ CCY CTG TAA AAA GGW CTA CAG CC 3ˊ) [38,39] and reverse primer H8121 (5ˊ GGG CAG CCR TGR ATT CAY TC 3ˊ) [40]. For each PCR sample, we used a total volume of 30 µL containing: 0.3 µL from each primer, 3 µL PCR buffer (Bioron GmbH, Ludwigshafen, Germany), 3 µL nucleotide mix (Bioron GmbH, Ludwigshafen, Germany), 0.2 µL Taq DNA Polymerase (Bioron GmbH, Ludwigshafen, Germany), 18.2 µL HPLC H2O (PanReac AppliChem, Darmstadt, Germany), and 5 µL template DNA. All samples were amplified in singleplex PCR, using a thermal cycler (SensoQuest GmbH, Göttingen, Germany) under the following conditions: 95 °C for 5 min as initial denaturation step, followed by 38 cycles of 95 °C for 45 s, 52 °C for 1 min, and 72 °C for 2 min. Final extension was done at 72 °C for 10 min and kept at 4 °C until removal from the thermal cycler. We verified the amplicons on 1.4% agarose gel and purified them following a standard ethanol precipitation protocol [36] or with Sephadex G-50 Fine solution (GE Healthcare, Uppsala, Sweden). For sequencing the Cytb gene we used the following primers: L14764 (see above) and MT-C2 (5ˊ TGA GGA CAA ATA TCA TTC TGA GG 3ˊ) [41], while for CO1 sequencing we used CO1-ExtF (5ˊ ACG CTT TAA CAC TCA GCC ATC TTA CC 3ˊ) [42] and Passer F1 (5ˊ CCA ACC ACA AAG ACA TCG GAA CC 3ˊ) [43]. All primers mentioned in this section were purchased from Eurofins Genomics, Ebersberg, Germany. Sanger sequencing was performed on an ABI 3739 Automated capillary sequencer by StarSEQ GmbH (Mainz, Germany).
2.3. Analysis of Mitochondrial Markers, Genetic Diversity, and Divergence Time
We assembled the raw sequences and visually inspected and corrected the alignments in CodonCode Aligner 5.01 (CodonCode Corporation, Centerville Massachusetts, USA). We trimmed all Cytb sequences to a fixed length of 909 base pairs and the CO1 to 642 base pairs. We used MEGA 7.0 [44] to check that our sequences contained no stop codons and that the open reading frame was identical for both Cytb and CO1 sequences. We concatenated these two genes in Seaview 4.7 [45] and performed all further analyses with the concatenated sequence. Upon verification, we employed DnaSP 6 [46] to check for nucleotide and haplotype diversity indexes and to group all samples from a certain country in a single population. Consequently, we analyzed the *arp file in Arlequin 3.5.2.2 [47] to obtain FST values. Via Arlequin software we also corrected the haplotype diversity values for the sample size.
To accurately identify the correct position of the nucleotide polymorphic sites in our trimmed sequences, we used two reference genes. For Cytb, we used a L. collurio complete Cytb sequence available in GenBank, accession number EF621589.1. However, for CO1 we could not find a complete sequence; we therefore used a L. isabellinus complete mitochondrial genome sample, GenBank accession number KP995437.1, from which we used the CO1 as a reference. We employed PopArt (Population Analysis with Reticulate Trees) [48] to visualize the haplotype network and geographic distribution across the western Palearctic.
To estimate evolutionary divergence time, we used the concatenated aligned sequences of our 132 RBS samples, to which we added 5 shrike species, with an identical alignment i.e., Cytb and CO1. We first retrieved from GenBank the complete mitochondrial genomes of the 5 species, after which we selected the CytB and CO1 and, in Seaview 4.7 we concatenated these two genes. From GenBank, we retrieved information for the following shrike species: L. schach (NC_030604.1), L. isabellinus (KP995437.1), L. cristatus (NC_028333.1), L. tephronotus (NC_028333.1), and L. sphenocercus (KU884610.1). As an outgroup, we used two corvid species, namely Corvus corax (KX245148.1) and Corvus frugilegus (Y18522.2), for which the alignment methodology was the same as mentioned above. We conducted the analyses in MEGA 7.0, using the Kimura 2-parameter model and modelled the rate variation among sites with a gamma distribution (shape parameter = 1). The analysis involved 139 nucleotide sequences i.e., 132 RBS, the 5 other Lanius species and the 2 Corvus species; each sequence had 1551 positions.
Finally, we performed all molecular dating with the programs BEAST 1.8.0 and BEAUTi 1.8.0 [49], using the CIPRES cyberinfrastructure for phylogenetic research [50]. To establish the divergence time, we applied the substitution rate of 2.1% substitutions/site/ million years for Cytb [51] and we estimated the rate for CO1. To select the most suitable evolutionary model, we used the jModeltest 2.1.10 software [52]. We further applied the HKY nucleotide substitution model, with a strict clock and Yule speciation process, a 106 chain length and sampling every 1000th tree. We then employed TreeAnnotator 1.8.0 [49] to summarize the sample of trees and FigTree 1.4.3 for final visualization [53].
3. Results
Our 1551-bp concatenated alignment revealed 76 haplotypes for the 132 RBS samples, with 0.96 haplotype diversity, 0.009 nucleotide diversity (π), and 110 segregating sites (Table 1). Taken separately, Cytb was characterized by 63 polymorphic sites and CO1 by 47 sites (Table S2). For the concatenated genes, we found the highest haplotype diversity (i.e., 1) in Bulgaria, France, Latvia and Romania (Table 1). The birds from Germany, Poland, Hungary, and the Czech Republic were characterized by a haplotype diversity of 0.974, 0.95, 0.934, and 0.857, respectively. The most common haplotype, haplotype 4, was represented by 23 individuals and it was spread across the entire breeding area (Figure 3 and Table S3). It was followed by haplotype 7, with a total of eight birds in Bulgaria, Romania, Spain, and Sweden. Haplotype 3 was represented by seven birds in Belgium, Germany, Latvia, and Spain. The next ten common haplotypes were distributed across 31 birds, with no apparent pattern. The remaining 63 haplotypes were singletons i.e., 63 RBS in our dataset were characterized by a unique haplotype.

Table 1.
Molecular diversity indexes for the 14 populations of breeding Red-backed Shrike (RBS), obtained from the concatenated genes cytochrome oxidase B (Cytb) and cytochrome c oxidase subunit 1 (CO1).
The pairwise FST values (Table 2) indicated low genetic differentiation between the majority of the 14 populations in our dataset. A certain level of differentiation was observed between shrikes from Czech Republic and The Netherlands, as well as between Russia and The Netherlands, and Russia and Ukraine. The value for Russia and Ukraine was most curious, due to their geographical position as neighboring countries and the absence of ecological barriers. Yet, the distance between the two-sampling locations was approximately 1160 km, which might explain the relatively low sharing of genetic material, in spite of their mobility and low philopatry rates. The haplotype network (Figure 1 and Figure 2) indicated a two-clade structure, with higher diversity in the larger clade. The two clades were separated by 26 mutation steps. The geographic distribution of haplotypes illustrates a higher number of singletons in France, Latvia, Hungary, and Romania (Figure 3 and Table S3).
Table 2.
Population pairwise FST indicators. Values which indicate low genetic differentiation (<0.05) are typed in bold. The rest of the values indicate moderate genetic differentiation (0.05 to 0.25). Units with significant p-values (<0.05) are marked with “+” above the diagonal.
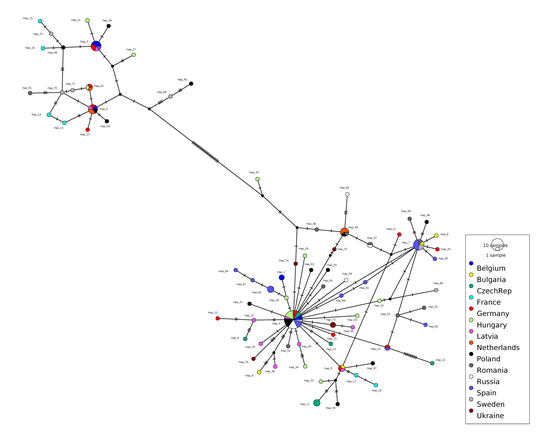
Figure 1.
Minimum-spanning haplotype network based on the concatenated Cytb and CO1 genes. Circle size reflects frequency of the haplotypes. The small black circles indicate missing node haplotypes. Each small vertical line traversing the connecting line between two haplotypes corresponds to one mutation step.
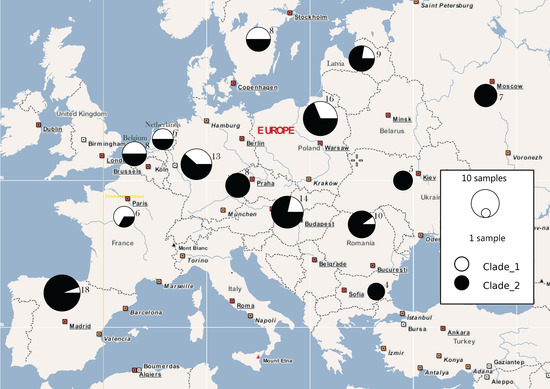
Figure 2.
Geographic distribution of the two main haplotype clades, based on concatenated Cytb and CO1, as represented by our mtDNA data from 132 birds in 14 countries. On the map, the unit near each circle indicates number of samples for the respective population.
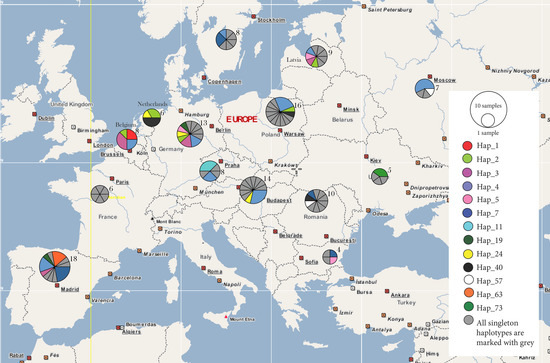
Figure 3.
Geographic distribution of the 76 haplotypes based on concatenated Cytb and CO1, as represented by our mtDNA data from 132 birds in 14 countries. On the right-side legend panel, each shared haplotype is assigned with a different color, whereas all singleton haplotypes are marked with grey. On the map, the unit near each circle indicates number of samples for the respective population.
Our phylogenetic tree (Figure 4, Appendix A, and Figure S1) and estimate of evolutionary divergence (Table 3) indicate a separation time between the two major clades of approximately 1.2 million years ago. Between L. collurio and its sister species—L. isabellinus, the divergence event is estimated at 1.9 million years before present. In comparison with L. cristatus, L. tephronotus, and L. schach, the RBS separated approximately 4.2 million years ago. In our analysis, L. sphenocercus scoreed the highest differentiation, separating itself from the other shrikes 6 million years ago. Regarding the outgroup, the two Corvus species, the divergence is estimated to have happened 12.3 million years before present.
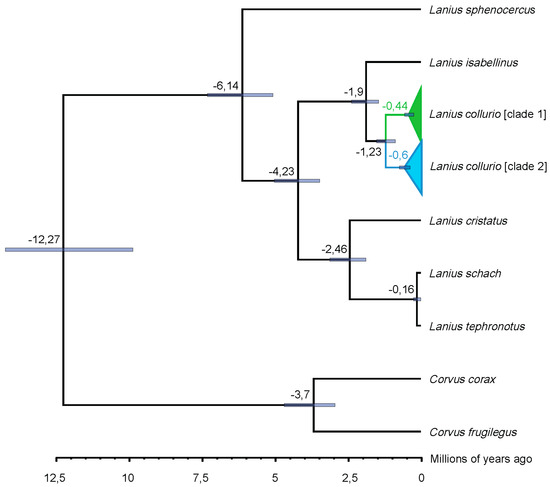
Figure 4.
Maximum clade credibility tree (Beast) based on Cytb and CO1 genes, for L. sphenocercus, L. isabellinus, L. collurio, L. cristatus, L. schach, L. tephronotus and, as outgroup, Corvus corax and Corvus frugilegus. Values expressed on branch ramifications indicate node age; 95% highest posterior densities are displayed as node bars.
Table 3.
Estimates of evolutionary divergence (base substitutions per site) over sequence pairs between the two main L. collurio clades, versus the other five Laniidae species and the Corvidae.
4. Discussion
4.1. Behind the Mask: Genetic Panmixia
Our mtDNA analysis provides the most complete view to date on the genetic structure of the RBSs breeding in the western Palearctic. The high number (n = 76), diversity index (0.96), and random distribution of haplotypes characterizing our 132 birds from 14 countries indicate significant genetic diversity and low evidence for population differentiation. This situation is not unique to the RBS, but actually a common phylogeographic status for many bird species currently distributed in Eurasia. Genetic admixture has been previously shown to characterize the Hoopoe (Upupa epops) population [54], the White-winged Snowfinch (Montifringila nivalis) [55], the Common Sandpiper (Actitis hypoleucos) [56], the Marsh Warbler (Acrocephalus palustris) [57], the Paddyfield Warbler (Acrocephalus agricola) [58], the Eurasian Reed Warbler (Acrocephalus scirpaceus) [59], the Western Capercaillie (Tetrao urogallus) [60], the Hazel Grouse (Tetrastes bonasia) [61], the Eurasian Blackcap (Sylvia atricapilla) [62], the European Bee-eater (Merops apiaster) [63], and the Eurasian Jay (Garrulus glandarius) [64], to name just a few. For the RBS, this genetic background is a clear result of Pleistocene climatic history (see next chapter), with different lineages coming together into refugia and, possibly, even bird movements between different refugia. We further speculate that, during the interglacial periods, lineages from different refugia came in contact at the suture zones, in the maximum population expansion times. Currently, this genetic diversity is under no evident selection pressure, due to their migratory behavior, which hinders sedentarism and population differentiation. Moreover, the RBS is characterized by low philopatry rates i.e., <10% [65,66] (plus Luís Reino, Franck Hollander, Piotr Tryjanowski, Marcin Tobółka, and Boris Nikolov pers. com.) and a wide continuous breeding range i.e., from Portugal until central Siberia [24], which further promotes gene flow among breeding populations. For European shrike species, random haplotype distribution has also been found in the Lesser Grey Shrike (L. minor) [18,67]. We also refer the readers to the Southern Grey Shrike (L. meridionalis koenigi) [20], which, although endemic to the Canary Islands and subject of a different population history (see next chapter), is likewise characterized by a high haplotype diversity i.e., 0.815 versus 0.96 in L. collurio (this study). Similarly, in non-European shrikes, the endemic island Loggerhead Shrike (L. ludovicianus anthonyi) indicates a random distribution of haplotypes across its restricted distribution in the northern California Chanel Islands [23]. In Africa, the Fiscal Shrike (L. collaris) is characterized by a haplotype diversity of 0.932, as shown by a study which found 26 haplotypes among 45 individuals [68].
In contrast, sedentary bird species in Europe, especially those with fragmented distribution, tend to show a better-defined genetic structure. To illustrate this, we mention the Black Grouse (Lyrurus tetrix), which overall shows specific haplotypes for each subpopulation in the Austrian Alps [69]. Regarding the distinct haplotype clades structure of the RBS, among European bird taxa, the occurrence of a two clade structure with no geographical distinction has been previously observed in the Common Redstart (Phoenicurus phoenicurus) [70] and the Eurasian Collared Dove (Streptopelia decaocto) [71]. For the two haplotype groups characterizing the Common Redstart, Johnsen et al. mention a high maximum intraspecific distance (%) i.e., 5.08 [42], whereas for the RBS the distance is 2.76. Additionally, in Eurasia, two haplotype clade structures have been observed in the Middle Spotted Woodpecker (Dendrocoptes medius), but with a clear differentiation between the European and Asian haplotypes [72]. A comparable situation has been indicated in the Little Owl (Athene noctua), with two distinctive clades for western and eastern European haplotypes [73]. Apparent western and eastern European haplotype clades have also been suggested for the Great Reed Warbler (Acrocephalus arundinaceus) [74]. For the Tawny Owl (Strix aluco), one genetic study has revealed the existence of three separate haplotype clades, one for each of the three main refugia (Iberian Peninsula, Italian Peninsula, and the Balkans) [75]. Similarly, on the North American continent, two distinct mitochondrial clades have been detected in the Northern Raven (Corvus corax) [76] and the Snow Goose (Anser caerulescens caerulescens) [77], and in both species a certain degree of geographic structuring of the clades is reported. Most commonly, high mtDNA divergence results from cryptic speciation, hybridization of closely related species or separation in isolated refugia during the glacial times. For the RBS, we suggest that the current situation is a result of the latter, as the molecular divergence time of the two clades coincides with the Pleistocene period. Nonetheless, because of limitations in our dataset and localities where samples were collected, we cannot exclude the possibility of introgressive hybridization with closely-related taxa or that we might have simply overlooked an extant lineage. Particularly for the RBS, a peculiar curiosity arises from the existence of its closest taxon, the Red-tailed Shrike (Lanius phoenicuroides). This species breeds at the western edge of the RBS areal and was previously considered a subspecies of the RBS, it is known to hybridize with the RBS and, its field identification provides great challenges due to morphological similarities. We believe the inclusion of Red-tailed Shrike samples in our RBS dataset might have offered crucial details regarding the two-clade structure, but at this stage we could not obtain a satisfactory number of samples. Nonetheless, we are confident that future analysis will shed light on this aspect.
Concerning the genetic diversity of country specific RBS populations in our study, in Belgium, Czech Republic, The Netherlands, and Russia there are less singletons and haplotype diversity is lowest (Figure 3, Table S3), which accounts for lower genetic diversity. Nonetheless, we mention that in our datasets per country, most samples were collected in the same region, simply because of collaborator’s availability. Relating to the RBS high genetic diversity across the studied populations, we underline two main hypotheses to explain this pattern: (i) Because our dataset revealed higher haplotype diversity in Bulgaria, Romania, France, and Latvia, we believe the glacial refugia for the European shrikes was in the Balkans, as for many other biota taxa [13]. The presence of France among the countries with the highest diversity in our dataset might also indicate a refugia in the Italian Peninsula, but unfortunately our data does not offer additional evidence in this direction and as such, it will be overconfident to make further statements. However, our diversity indexes per country generally imply that, the further away the RBS population expanded, the lower the genetic diversity [7,30]. Additionally, all RBS from western Europe migrate to Africa via the Balkans [65,78,79], even the birds which breed in the Iberian Peninsula [80]. The former information, correlated with our genetic results, indicates a clear population expansion pattern from the Balkans towards western Europe. (ii) Secondly, for Belgium and The Netherlands, this lower genetic diversity can be attributed to population crashes in the second half of last century, mainly as a response of habitat loss and intensive farming practices. In Belgium, for example, in the 1960’s the population crashed to only 600 pairs from the original numbers of about 5000 pairs [81], but recent counts indicate a total population of 3700 pairs, distributed only in Wallonia (in Flanders there are only 1 to 5 RBS pairs) [82]. Therefore, our results suggest that the RBS population in western Europe is more vulnerable, in terms of population conservation. Nonetheless, we mention Spain, where although the RBS is suffering a steady decline, with some local populations having dropped by 95% [83,84], genetic diversity is still high. Yet, from a species conservation perspective, the current prevailing threat for the RBS comes from the hunting of migratory birds in the Mediterranean countries [27,85,86,87], where, recent data revealed that in Egypt alone more than 30,000 RBS are taken each autumn [28].
To conclude, we emphasize the high genetic diversity for the breeding populations included in our study, the characteristic two-clade haplotype structure, and the apparent gene flow, which is reassuring when taking into account the hunting of thousands of RBS in the Mediterranean basin, the alarming rate of habitat loss across Europe and the climatic changes occurring along the migratory pathways.
4.2. A Blast from the Past: Pleistocene Upbringing
Population structure is determined by the genotype distribution in space and time and is the result of past events as well as ongoing processes [88]. Across the western Palearctic, the genetic population structure for majority of taxa has been shaped by the complex climatic shifts, vegetation composition, and refugium locations in the Quaternary [13]. For many species of plants, amphibians, reptiles, birds, and mammals, this translates into genetic admixture, triggered by the numerous glaciations which forced populations to retreat to refugia, where the fusion of different groups of individuals took place; subsequently, genetic admixture was reinforced when breeding populations from different glacial refugia fused. Overall, for plants species and animals with limited mobility, some population differentiation has occurred, and individuals can be grouped in clades belonging to their current geographic location or clades belonging to certain refugia, with the highest diversity concentrated near the glacial refugia e.g., Balkans, as observed in our RBS study as well. The Black Alder (Alnus glutinosa) [89] and oak trees (genus Quercus) [90] have also spread to North Europe from the Balkan Peninsula. Schrimpf et al. revealed that South-east Europe is the hotspot for genetic diversity of the Noble Crayfish (Astacus astacus) [91]. In the European Fire-bellied Toads (Bombina bombina & Bombina variegata), nuclear and mitochondrial phylogeography indicate region specific clades across their breeding range [92]. For reptiles, the European Pond Turtle (Emys orbicularis) shows high intraspecific differentiation, with haplotypes limited to certain geographic areas [93]. Furthermore, Whipsnakes also show population differentiation and it appears that they colonized Europe from the Balkan refugia [94,95]. Wall Lizard genetic population structure likewise indicate the Balkans as refugia and diversity hotspot [96]. Among mammals, the Brown Bear (Ursus arctos) haplotypes can be divided in western and eastern clades, but only Romanian bears belong to both clades [97], giving further evidence of the high genetic diversity which occurs in South-east Europe.
Whereas in the northern temperate regions, many taxa show genetic admixture caused by Pleistocene glaciations and recent expansion from the refugia, across the tropical areas, bird species, especially the sedentary ones, show a clearly differentiated structure, with haplotypes being safely assigned to certain regions. We mention the African Blue Tits (Cyanistes teneriffae) in the Canary Islands, which belong to six haplotype groups, one for each of the seven islands in the archipelago (Lanzarote having just one haplotype) [98]. For the South American continent, a recent study revealed that Polioptila gnatcatchers have a remarkable phylogeographic differentiation, mainly attributed to the landscape changes which occurred during the late Pliocene [99].
In conclusion, the RBS mitochondrial phylogeography indicates genetic admixture among the sampled populations, a common feature for bird species inhabiting the western Palearctic. Moreover, for RBS, we recorded higher genetic diversity in South-east Europe, further consolidating the theory that, during the Pleistocene glaciations, the biggest refugia for this species was in the Balkan Peninsula. From this refugia, the species has subsequently expanded and colonized the whole of Europe.
5. Conclusions
To the best of our knowledge, this study is the first to shed light upon the genetic population structure of RBSs. Across the western Palearctic, breeding RBSs show high genetic diversity, with a total of 76 haplotypes and 0.96 haplotype diversity in our dataset from 132 birds in 14 countries. To our surprise, two major lineages are apparent, with no geographic distinction, similar to the situation of the Common Redstart and the Eurasian Collared Dove. The random geographic distribution of haplotypes, a general indicator for population admixture, is common and widespread among European biota. This situation originates in the Pleistocene era, whose climate was marked by dramatic oscillations between periods of extreme cold and extensive ice coverage and periods of relative warmth. These climatic rotations, which implied a ‘back-and-forth’ process of population expansion and subsequent retreat to refugia, represent the driving factor that shaped today’s genetic admixture in RBS across their breeding range in the western Palearctic. In future research, we will try to elucidate the evolutionary past of the RBS by using genomic next generation sequencing analyses, which allow a much higher resolution than mtDNA sequences [100].
Supplementary Materials
The following are available online at https://www.mdpi.com/1424-2818/11/3/31/s1, Video S1: How to collect buccal mucosa from a live bird, using a cotton swab—A video protocol. Table S1: Complete details of used samples. Table S2: Polymorphic sites, positions and haplotype frequency. Table S3: Haplotype distribution per country. Figure S1: Laniidae and Corvidae Phylogenetic Tree.
Author Contributions
M.W. conceived and designed the study. L.G.P. performed the field and laboratory work. M.W. contributed reagents, materials and analysis tools. L.G.P. and R.C.F.-S. analyzed the data. L.G.P. wrote the manuscript, with regular intellectual input from R.C.F.-S. and M.W.
Funding
The study was supported by grants from the Deutsche Ornithologen-Gessellschaft (DO-G)–Abs-Halbstipendium, the Erwin-Stresemann Förderung and the Irmgard und Michael Abs Stiftung to L.G.P.
Acknowledgments
Our sincere appreciation goes to Erjia Wang, Carina Carneiro de Melo Moura, Beate Weibel and Dieter Thomas Tietze for providing useful insights on the topic and initiating numerous though-provoking discussions. We also acknowledge Hedwig Sauer-Gürth, Heidi Staudter and Petra Fellhauer for technical support. We are indebted to Grigor Jorgo (Albania), Franck Hollander (Belgium), Rien van Wijk (Bulgaria), Sharina van Boheemen (Czech Republic), Riho Marja and Jaanus Elts (Estonia), Bussière Raphaël (France), Markus Santhosh Braun, Jan Engler, Christoph Unger, Jens Hering, Fabian Schrauth, Silke Fregin and the Zoological Institute and Museum at the Ernst-Moritz-Arndt University of Greifswald, Simon Bruslund and Heidelberg Zoo (Germany), András Vizkert, Norbert Mátrai, Zsolt Karcza and Bianka Regina Jabsa (Hungary), Yariv Malihi, Yoav Motro, Noam Weiss and the International Birding and Research Center Eilat (Israel), Oskars Keišs and Valts Jaunzemis (Latvia), Stef Waasdorp (The Netherlands), Arild Johnsen, Lars Erik Johannessen and the Natural History Museum at University of Oslo (Norway), Marcin Tobółka and Piotr Tryjanowski (Poland), Peter Pap Laszlo and Csongor Istvan Vágási (Romania), Tatiana Makarova, Ivan Starikov and the Zoological Museum of Moscow University (Russia), Craig Symes (South Africa), Lluís Mir Pla, Oriol Baltà, Pere Josa Anguera and Gerard Dalmau Bonet (Spain), Ulf Johansson and the Swedish National History Museum (Sweden) for providing the samples used in our study. We also acknowledge Lykke Pedersen for useful advice on how to trap wild Red-backed Shrikes. We further thank Márton Zsoldos and Dan S. Bandacu for allowing us to use their Red-backed Shrike illustrations and photos at various stages of the study. Mare Haider, Pelin and Irmgard Yildiz, Philipp Scheffzek and Markus Schirmer provided necessary support at the incipient stage of the study. Moreover, we acknowledge the Institute for Avian Research “Vogelwarte Helgoland” in Wilhelmshaven and Franz Bairlein for help during the editing of this manuscript. We thank Kylynn Clare (International Birding and Research Center Eilat) for improving the language and grammar of the manuscript. Fieldwork in Germany was approved by the Vogelwarte Radolfzell. We acknowledge the Carl Zeiss Sports Optics GmbH for providing L.G.P. with a “Victory SF” 10 × 42 binoculars for fieldwork, and Petzl Deutschland GmbH for providing a “Reactik +” headlamp. We thank the six anonymous reviewers for their constructive comments which helped to improve a previous version of the manuscript. Finally, we dedicate this research article to Dietlinde Schiebel (Rohrbach, Heidelberg), with whom L.G.P. had fruitful ornithological discussions; she passed away in summer of 2017 following a short, severe illness.
Conflicts of Interest
Michael Wink is Editor-in-Chief of the Diversity journal. The funding agencies had no role in the collection, analysis or interpretation of the data, nor in the writing of the manuscript and decision to publish the results.
Appendix A
Maximum clade credibility tree (Beast) based on Cytb and CO1 genes, for L. sphenocercus, L. isabellinus, L. collurio, L. cristatus, L. schach, L. tephronotus and, as outgroup, Corvus corax and Corvus frugilegus.

References
- Hewitt, G. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.R.; Lister, A.M.; Barnes, I.; Dalén, L. Refugia revisited: Individualistic responses of species in space and time. Proc. R. Soc. B Biol. Sci. 2010, 277, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Huybers, P. Glacial variability over the last two million years: An extended depth-derived agemodel, continuous obliquity pacing, and the Pleistocene progression. Quat. Sci. Rev. 2007, 26, 37–55. [Google Scholar] [CrossRef]
- Lisiecki, L.E.; Raymo, M.E. Plio-Pleistocene climate evolution: Trends and transitions in glacial cycle dynamics. Quat. Sci. Rev. 2007, 26, 56–69. [Google Scholar] [CrossRef]
- Pisias, N.G.; Moore, T.C. The evolution of Pleistocene climate: A time series approach. Earth Planet. Sci. Lett. 1981, 52, 450–458. [Google Scholar] [CrossRef]
- Hewitt, G.M. Speciation, hybrid zones and phylogeography—Or seeing genes in space and time. Mol. Ecol. 2001, 10, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Médail, F.; Diadema, K. Glacial refugia influence plant diversity patterns in the Mediterranean Basin. J. Biogeogr. 2009, 36, 1333–1345. [Google Scholar] [CrossRef]
- Hewitt, G.M. Post-glacial re-colonization of European biota. Biol. J. Linn. Soc. 1999, 68, 87–112. [Google Scholar] [CrossRef]
- Černa Bolfíková, B.; Eliášová, K.; Loudová, M.; Kryštufek, B.; Lymberakis, P.; Sándor, A.D.; Hulva, P. Glacial allopatry vs. postglacial parapatry and peripatry: The case of hedgehogs. PeerJ 2017, 5, e3163. [Google Scholar] [CrossRef] [PubMed]
- Webb, T., III. Glacial and Holocene vegetation history: Eastern North America. In Handbook of Vegetation Science; Huntley, B., Webb, T., III, Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1988; pp. 385–414. [Google Scholar]
- Taberlet, P.; Fumagalli, L.; Wust-Saucy, A.G.; Cosson, J.F. Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol. 1998, 7, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Remington, C.L. Suture-Zones of Hybrid Interaction Between Recently Joined Biotas. In Evolutionary Biology: Volume 2; Dobzhansky, T., Hecht, M.K., Steere, W.C., Eds.; Springer US: Boston, MA, USA, 1968; pp. 321–428. ISBN 978-1-4684-8094-8. [Google Scholar]
- Weiss, S.; Ferrand, N. Phylogeography of Southern European Refugia; Springer: Dordrecht, The Netherlands, 2007; ISBN 978-1-4020-4903-3. [Google Scholar]
- Avise, J.C. Evolutionary Pathways in Nature: A Phylogenetic Approach; Cambridge University Press: Cambridge, UK, 2006; ISBN 9780511606939. [Google Scholar]
- Wiley, E.O.; Lieberman, B.S. Phylogenetics: Theory and Practice of Phylogenetic Systematics, 2nd ed.; Willey Blackwell: Hoboken, NJ, USA, 2011; ISBN 9780470905968. [Google Scholar]
- Del Hoyo, J.; Collar, N.J. Illustrated Checklist of the Birds of the World, Volume 2: Passerines; Lynx Edicions: Barcelona, Spain, 2016; ISBN 978-84-96553-98-9. [Google Scholar]
- Gill, F.; Donsker, D. IOC World Bird List v8.1. Available online: http://www.worldbirdnames.org/ (accessed on 16 February 2018).
- Kvist, L.; Giralt, D.; Valera, F.; Hoi, H.; Kristin, A.; Darchiashvili, G.; Lovaszi, P. Population decline is accompanied by loss of genetic diversity in the Lesser Grey Shrike Lanius minor. Ibis (Lond. 1859) 2011, 153, 98–109. [Google Scholar] [CrossRef]
- Olsson, U.; Alström, P.; Svensson, L.; Aliabadian, M.; Sundberg, P. The Lanius excubitor (Aves, Passeriformes) conundrum-Taxonomic dilemma when molecular and non-molecular data tell different stories. Mol. Phylogenet. Evol. 2010, 55, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Padilla, D.P.; Spurgin, L.G.; Fairfield, E.A.; Illera, J.C.; Richardson, D.S. Population history, gene flow, and bottlenecks in island populations of a secondary seed disperser, the southern grey shrike (Lanius meridionalis koenigi). Ecol. Evol. 2015, 5, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.; Wink, M.; Garcia-del-Rey, E.; Delgado Castro, G. Evidence from DNA nucleotide sequences and ISSR profiles indicates paraphyly in subspecies of the Southern Grey Shrike (Lanius meridionalis). J. Ornithol. 2008, 149, 495–506. [Google Scholar] [CrossRef]
- Mundy, N.I.; Winchell, C.S.; Woodruff, D.S. Genetic differences between the endangered San Clemente Island loggerhead shrike Lanius ludovicianus mearnsi and two neighbouring subspecies demonstrated by mtDNA control region and cytochrome b sequence variation. Mol. Ecol. 1997, 6, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Caballero, I.C.; Ashley, M.V. Genetic analysis of the endemic island loggerhead shrike, Lanius ludovicianus anthonyi. Conserv. Genet. 2011, 12, 1485–1493. [Google Scholar] [CrossRef]
- del Hoyo, J.; Elliott, A.; Christie, D.A. (Eds.) Handbook of the Birds of the World. Vol. 13. Penduline-Tits to Shrikes; Lynx Edicions: Barcelona, Spain, 2008. [Google Scholar]
- Euro Bird Portal. Available online: http://www.eurobirdportal.org/ebp/en/#home/LANCOL/r2000/LANTOR/r2000/ (accessed on 1 March 2018).
- IUCN Red List. Available online: https://www.iucnredlist.org/species/22705001/110988087 (accessed on 31 October 2018).
- Brochet, A.-L.; Van den Bosschen, W.; Jbour, S.; Ndang’ang’a, P.K.; Jones, V.R.; Abdou, W.A.L.I.; Al-hmoud, A.R.; Asswad, N.G.; Atienza, J.C.; Atrash, I.; et al. Preliminary assessment of the scope and scale of illegal killing and taking of birds in the Mediterranean. Bird Conserv. Int. 2016, 26, 1–28. [Google Scholar] [CrossRef]
- Eason, P.; Rabia, B.; Attum, O. Hunting of migratory birds in North Sinai, Egypt. Bird Conserv. Int. 2016, 26, 39–51. [Google Scholar] [CrossRef]
- Allendorf, F.W.; Luikart, G.; Aitken, S.N. Conservation and the Genetics of Populations, 2nd ed.; Willey Blackwell: Hoboken, NJ, USA, 2012. [Google Scholar]
- Hartl, D.L.; Clark, A.G. Principles of Population Genetics, 3rd ed.; Sinauer Associates, Inc.: Sunderland, MA, USA, 1997; ISBN 0-87893-308-5. [Google Scholar]
- Krebs, J.E.; Goldstein, E.S.; Kilpatrick, S.T. Lewin’s Genes XII; Jones & Bartlett Learning: Burlington, MA, USA, 2017; ISBN 9780763766320. [Google Scholar]
- Mindell, D.P. (Ed.) Avian Molecular Evolution and Systematics; Academic Press: London, UK, 1997. [Google Scholar]
- Rubinoff, D.; Holland, B.S. Between two extremes: Mitochondrial DNA is neither the panacea nor the nemesis of phylogenetic and taxonomic inference. Syst. Biol. 2005, 54, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Hurst, G.D.D.; Jiggins, F.M. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: The effects of inherited symbionts. Proc. R. Soc. B Biol. Sci. 2005, 272, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, F.; Alexandrino, J.; Weiss, S.; Ferrand, N. Documenting the advantages and limitations of different classes of molecular markers in a well-established phylogeographic context: Lessons from the Iberian endemic Golden-striped salamander, Chioglossa lusitanica (Caudata: Salamandridae). Biol. J. Linn. Soc. 2008, 95, 371–387. [Google Scholar] [CrossRef]
- Green, M.R.; Sambrook, J. Molecular Cloning: A Laboratory Manual, Vol. 1, 4th ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2012. [Google Scholar]
- Sorenson, M.D.; Ast, J.C.; Dimcheff, D.E.; Yuri, T.; Mindell, D.P. Primers for a PCR-based approach to mitochondrial genome sequencing in birds and other vertebrates. Mol. Phylogenet. Evol. 1999, 12, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, M.D.; Quinn, T.W. Numts: A Challenge for Avian Systematics and Population Biology. Auk 1998, 115, 214–221. [Google Scholar] [CrossRef]
- Miranda, H.C.; Kennedy, R.S.; Mindell, D.P. Phylogenetic placement of Mimizuku gurneyi (Aves: Strigidae) inferred from mitochondrial DNA. Auk 1997, 114, 315–323. [Google Scholar] [CrossRef]
- Sorenson, M.D. Avian mtDNA Primers; Boston University: Boston, MA, USA, 2003. [Google Scholar]
- Wink, M.; Sauer-Gürth, H.; Heidrich, P.; Witt, H.-H.; Gwinner, E. A Molecular Phylogeny of Stonechats and Related Turdids. In A Guide to the Genus Saxicola; Christopher Helm: London, UK, 2002; pp. 22–30. [Google Scholar]
- Johnsen, A.; Rindal, E.; Ericson, P.G.P.; Zuccon, D.; Kerr, K.C.R.; Stoeckle, M.Y.; Lifjeld, J.T. DNA barcoding of Scandinavian birds reveals divergent lineages in trans-Atlantic species. J. Ornithol. 2010, 151, 565–578. [Google Scholar] [CrossRef]
- Lohman, D.J.; Prawiradilaga, D.M.; Meier, R. Improved COI barcoding primers for Southeast Asian perching birds (Aves: Passeriformes). Mol. Ecol. Resour. 2009, 9, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Guindon, S.; Gascuel, O. Sea view version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar] [CrossRef]
- Weir, J.T.; Schluter, D. Calibrating the avian molecular clock. Mol. Ecol. 2008, 17, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A. FigTree v1. 3.1; Institute of Evolutionary Biology, University of Edinburgh: Edinburgh, UK, 2010. [Google Scholar]
- Wang, E.; van Wijk, R.E.; Braun, M.S.; Wink, M. Gene flow and genetic drift contribute to high genetic diversity with low phylogeographical structure in European Hoopoes (Upupa epops). Mol. Phylogenet. Evol. 2017, 113, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Resano-Mayor, J.; Fernández-Martín, Á.; Hernández-Gómez, S.; Toranzo, I.; España, A.; Gil, J.A.; de Gabriel, M.; Roa-Álvarez, I.; Strinella, E.; Hobson, K.A.; et al. Integrating genetic and stable isotope analyses to infer the population structure of the White-winged Snowfinch Montifringilla nivalis in Western Europe. J. Ornithol. 2017, 158, 395–405. [Google Scholar] [CrossRef]
- Zink, R.M.; Pavlova, A.; Drovetski, S.; Rohwer, S. Mitochondrial phylogeographies of five widespread Eurasian bird species. J. Ornithol. 2008, 149, 399–413. [Google Scholar] [CrossRef]
- Arbabi, T.; Gonzalez, J.; Wink, M. Mitochondrial evidence for genetic diversity and low phylogeographic differentiation in the Marsh Warbler Acrocephalus palustris (Aves: Acrocephalidae). Org. Divers. Evol. 2014, 14, 409–417. [Google Scholar] [CrossRef]
- Zehtindjiev, P.; Ilieva, M.; Hansson, B.; Oparina, O.; Oparin, M.; Bensch, S. Population genetic structure in the paddyfield warbler (Acrocephalus agricola Jerd.). Curr. Zool. 2011, 57, 63–71. [Google Scholar] [CrossRef]
- Arbabi, T.; Gonzalez, J.; Witt, H.H.; Klein, R.; Wink, M. Mitochondrial phylogeography of the Eurasian Reed Warbler Acrocephalus scirpaceus and the first genetic record of A. s. fuscus in Central Europe. Ibis (Lond. 1859) 2014, 156, 799–811. [Google Scholar] [CrossRef]
- Rutkowski, R.; Zawadzka, D.; Merta, D.; Stanković, A.; Jagołkowska, P.; Suchecka, E.; Kobielski, J. Conservation genetics of the Capercaillie Tetrao urogallus in Poland—Diversity of mitochondrial DNA in remnant and extinct populations. Acta Ornithol. 2017, 52, 179–196. [Google Scholar] [CrossRef]
- Rutkowski, R.; Jagołkowska, P.; Zawadzka, D.; Bogdanowicz, W. Impacts of forest fragmentation and post-glacial colonization on the distribution of genetic diversity in the Polish population of the hazel grouse Terastes bonasia. Eur. J. Wildl. Res. 2016, 62, 293–306. [Google Scholar] [CrossRef]
- Perez-Tris, J.; Bensch, S.; Carbonell, R.; Helbig, A.J.; Telleria, J.L. Historical diversification of migration patterns in a passerine bird. Evolution (N. Y.) 2004, 58, 1819–1832. [Google Scholar] [CrossRef]
- Carneiro de Melo Moura, C.; Bastian, H.-V.; Bastian, A.; Wang, E.; Wang, X.; Wink, M. Pliocene Origin, Ice Ages and Postglacial Population Expansion Have Influenced a Panmictic Phylogeography of the European Bee-Eater. Diversity 2019, 11, 12. [Google Scholar] [CrossRef]
- Aoki, D.; Kinoshita, G.; Kryukov, A.P.; Nishiumi, I.; Lee, S.; Suzuki, H. Quaternary-related genetic differentiation and parallel population dynamics of the Eurasian Jay (Garrulus glandarius) in the circum-Japan Sea region. J. Ornithol. 2018, 159, 1087–1097. [Google Scholar] [CrossRef]
- Cramp, S.; Perrins, C.M. (Eds.) The Birds of the Western Palearctic; Oxford University Press: Oxford, UK, 1993. [Google Scholar]
- Tryjanowski, P.; Goławski, A.; Kuźniak, S.; Mokwa, T.; Antczak, M. Disperse or stay? Exceptionally high breeding-site infidelity in the red-backed shrike Lanius collurio. Ardea 2007, 95, 316–320. [Google Scholar] [CrossRef]
- Giralt, D. Decline of the Lesser Grey Shrike (Lanius minor) at the Western Limit of the Distribution Area: Causes, Mechanisms and Conservation Proposals; University of Barcelona: Barcelona, Spain, 2015. [Google Scholar]
- Fuchs, J.; Crowe, T.M.; Bowie, R.C.K. Phylogeography of the fiscal shrike (Lanius collaris): A novel pattern of genetic structure across the arid zones and savannas of Africa. J. Biogeogr. 2011, 38, 2210–2222. [Google Scholar] [CrossRef]
- Sittenthaler, M.; Kunz, F.; Szymusik, A.; Grünschachner-Berger, V.; Krumböck, S.; Stauffer, C.; Nopp-Mayr, U. Fine-scale genetic structure in an eastern Alpine black grouse Tetrao tetrix metapopulation. J. Avian Biol. 2018, 49, 1–14. [Google Scholar] [CrossRef]
- Hogner, S.; Laskemoen, T.; Lifjeld, J.T.; Porkert, J.; Kleven, O.; Albayrak, T.; Kabasakal, B.; Johnsen, A. Deep sympatric mitochondrial divergence without reproductive isolation in the common redstart Phoenicurus phoenicurus. Ecol. Evol. 2012, 2, 2974–2988. [Google Scholar] [CrossRef] [PubMed]
- Bagi, Z.; Dimopoulos, E.A.; Loukovitis, D.; Eraud, C.; Kusza, S. MtDNA genetic diversity and structure of Eurasian Collared Dove (Streptopelia decaocto). PLoS ONE 2018, 13, e0193935. [Google Scholar] [CrossRef] [PubMed]
- Kamp, L.; Pasinelli, G.; Milanesi, P.; Drovetski, S.V.; Kosiński, Z.; Kossenko, S.; Robles, H.; Schweizer, M. Significant Asia-Europe divergence in the middle spotted woodpecker (Aves, Picidae). Zool. Scr. 2018, 1–16. [Google Scholar] [CrossRef]
- Pellegrino, I.; Negri, A.; Cucco, M.; Mucci, N.; Pavia, M.; Šálek, M.; Boano, G.; Randi, E. Phylogeography and Pleistocene refugia of the Little Owl Athene noctua inferred from mtDNA sequence data. Ibis (Lond. 1859) 2014, 156, 639–657. [Google Scholar] [CrossRef]
- Hansson, B.; Hasselquist, D.; Tarka, M.; Zehtindjiev, P.; Bensch, S. Postglacial colonisation patterns and the role of isolation and expansion in driving diversification in a passerine bird. PLoS ONE 2008, 3, e2794. [Google Scholar] [CrossRef] [PubMed]
- Brito, P.H. The influence of Pleistocene glacial refugia on tawny owl genetic diversity and phylogeography in Western Europe. Mol. Ecol. 2005, 14, 3077–3094. [Google Scholar] [CrossRef] [PubMed]
- Webb, W.C.; Marzluff, J.M.; Omland, K.E. Random interbreeding between cryptic lineages of the Common Raven: Evidence for speciation in reverse. Mol. Ecol. 2011, 20, 2390–2402. [Google Scholar] [CrossRef] [PubMed]
- Quinn, T.W. The genetic legacy of Mother Goose—Phylogeographic patterns of lesser snow goose Chen caerulescens caerulescens maternal lineages. Mol. Ecol. 1992, 1, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Tottrup, A.P.; Klaassen, R.H.G.; Strandberg, R.; Thorup, K.; Kristensen, M.W.; Jorgensen, P.S.; Fox, J.; Afanasyev, V.; Rahbek, C.; Alerstam, T. The annual cycle of a trans-equatorial Eurasian-African passerine migrant: Different spatio-temporal strategies for autumn and spring migration. Proc. R. Soc. B Biol. Sci. 2012, 279, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.; Fraser, K.C.; Kyser, T.K.; Tøttrup, A.P. Combining direct and indirect tracking techniques to assess the impact of sub-Saharan conditions on cross-continental songbird migration. J. Ornithol. 2016, 157, 1037–1047. [Google Scholar] [CrossRef]
- Tøttrup, A.P.; Pedersen, L.; Onrubia, A.; Klaassen, R.H.G.; Thorup, K. SM Migration of red-backed shrikes from the Iberian Peninsula: Optimal or sub-optimal detour? J. Avian Biol. 2017, 48, 149–154. [Google Scholar] [CrossRef]
- Titeux, N.; Van der Elst, D.; Van Nieuwenhuyse, D. Pie-grièche écorcheur, Lanius collurio. In Atlas Des Oiseaux Nicheurs de Wallonie 2001–2007; Jacob, J.-P., Dehem, C., Burnel, A., Dambiermont, J.-L., Fasol, M., Kinet, T., Van der Elst, D., Paquet, J.-Y., Eds.; Aves & Région Wallonne: Gembloux, Belgium, 2010; pp. 396–397. [Google Scholar]
- Derouaux, A.; Paquet, J.-Y. The worrying trends in breeding bird populations of Wallonia: 28 years of common bird monitoring. Bull. Aves 2018, 55, 1–31. [Google Scholar]
- Tellería, J.L. Distribution of the Red-Backed Shrike Lanius Collurio at Its Western Range Boundary: Patterns and Conservation Prospects. Ardeola 2018, 65, 221–232. [Google Scholar] [CrossRef]
- Tellería, J.L. Old Counts Suggest the Collapse of Two Red-Backed Shrike Lanius collurio Populations. Ardeola 2018, 65, 283–290. [Google Scholar] [CrossRef]
- Birdlife International. The Killing; Birdlife International: Cambridge, UK, 2015. [Google Scholar]
- Birdlife International. The Killing 2.0. A view to Kill; Birdlife International: Cambridge, UK, 2017. [Google Scholar]
- Hirschfeld, A.; Attard, G. Vogeljagd in Europa—Analyse von abschusszahlen und auswirkungen der jagd auf den erhalt bedrohter Vogelarten. Vogelschutz 2017, 53, 15–42. [Google Scholar]
- Hewitt, G.M.; Butlin, R.K. Causes and consequences of population structure. In Behavioural Ecology: An Evolutionary Approach; Krebs, J.R., Davies, N.B., Eds.; Blackwell: Oxford, UK, 1997; pp. 350–372. [Google Scholar]
- King, R.A.; Ferris, C. Chloroplast DNA phylogeography of Alnus glutinosa (L.) Gaertn. Mol. Ecol. 1998, 7, 1151–1161. [Google Scholar] [CrossRef]
- Brewer, S.; Cheddadi, R.; de Beaulieu, J.L.; Reille, M. The spread of deciduous Quercus throughout Europe since the last glacial period. For. Ecol. Manag. 2002, 156, 27–48. [Google Scholar] [CrossRef]
- Schrimpf, A.; Theissinger, K.; Dahlem, J.; Maguire, I.; Pârvulescu, L.; Schulz, H.K.; Schulz, R. Phylogeography of noble crayfish (Astacus astacus) reveals multiple refugia. Freshw. Biol. 2014, 59, 761–776. [Google Scholar] [CrossRef]
- Fijarczyk, A.; Nadachowska, K.; Hofman, S.; Litvinchuk, S.N.; Babik, W.; Stuglik, M.; Gollmann, G.; Choleva, L.; Cogǎlniceanu, D.; Vukov, T.; et al. Nuclear and mitochondrial phylogeography of the European fire-bellied toads Bombina bombina and Bombina variegata supports their independent histories. Mol. Ecol. 2011, 20, 3381–3398. [Google Scholar] [CrossRef] [PubMed]
- Lenk, P.; Fritz, U.; Joger, U.; Wink, M. Mitochondrial phylogeography of the European pond turtle, Emys orbicularis (Linnaeus 1758). Mol. Ecol. 1999, 8, 1911–1922. [Google Scholar] [CrossRef] [PubMed]
- Mezzasalma, M.; Di Febbraro, M.; Guarino, F.M.; Odierna, G.; Russo, D. Cold-blooded in the Ice Age: “refugia within refugia”, inter-and intraspecific biogeographic diversification of European whipsnakes (Squamata, Colubridae, Hierophis). Zoology 2018. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.T.; Bellaagh, M.; Wink, M.; Paunović, A.; Korsós, Z. Phylogeography of the Caspian whipsnake in Europe with emphasis on the westernmost populations. Amphib. Reptil. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Psonis, N.; Antoniou, A.; Karameta, E.; Leaché, A.D.; Kotsakiozi, P.; Darriba, D.; Kozlov, A.; Stamatakis, A.; Poursanidis, D.; Kukushkin, O.; et al. Resolving complex phylogeographic patterns in the Balkan Peninsula using closely related wall-lizard species as a model system. Mol. Phylogenet. Evol. 2018, 125, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Kohn, M.; Knauer, F.; Stoffella, A.; Schröder, W.; Pääbo, S. Conservation genetics of the European brown bear—A study using excremental PCR of nuclear and mitochondrial sequences. Mol. Ecol. 1995, 4, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Dietzen, C.; Garcia-del-Rey, E.; Castro, G.D.; Wink, M. Phylogeography of the blue tit (Parus teneriffae-group) on the Canary Islands based on mitochondrial DNA sequence data and morphometrics. J. Ornithol. 2008, 149, 1–12. [Google Scholar] [CrossRef]
- Carneiro de Melo Moura, C.; de Araujo, H.F.; Aleixo, A.; Wink, M.; Fernandes, A.M. The role of landscape change and paleoclimatic events in shaping the evolutionary history of the Polioptila gnatcatchers (Passeriformes, Polioptilidae) with emphasis on species associated with open habitats. J. Avian Biol. 2018, 49, 1–12. [Google Scholar] [CrossRef]
- Kraus, R.H.S.; Wink, M. Avian genomics: Fledging into the wild! J. Ornithol. 2015, 156, 851–865. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).