High Genetic Diversity among Breeding Red-Backed Shrikes Lanius collurio in the Western Palearctic



Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. DNA Extraction, Polymerase Chain Reaction, and Sequencing
2.3. Analysis of Mitochondrial Markers, Genetic Diversity, and Divergence Time
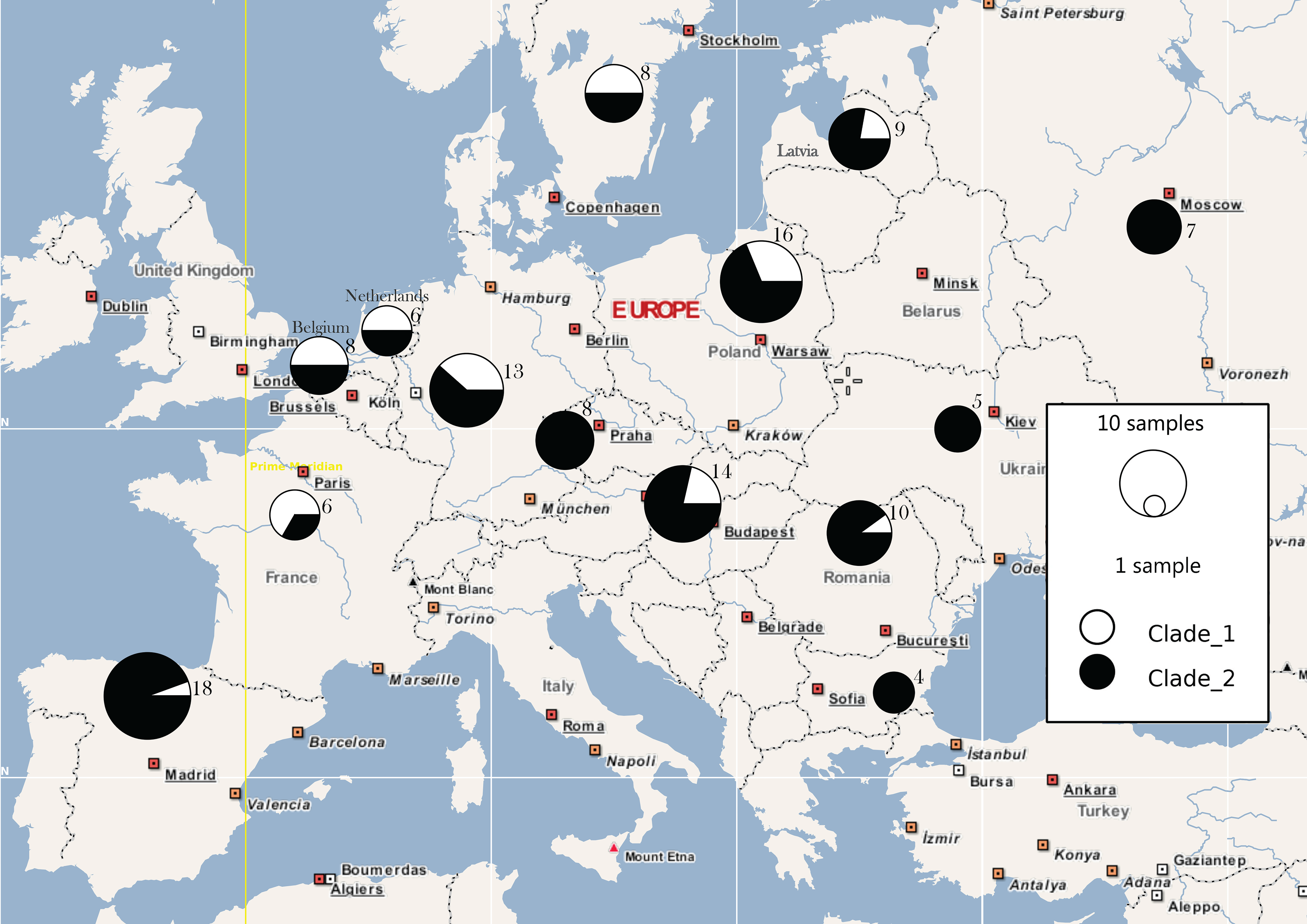
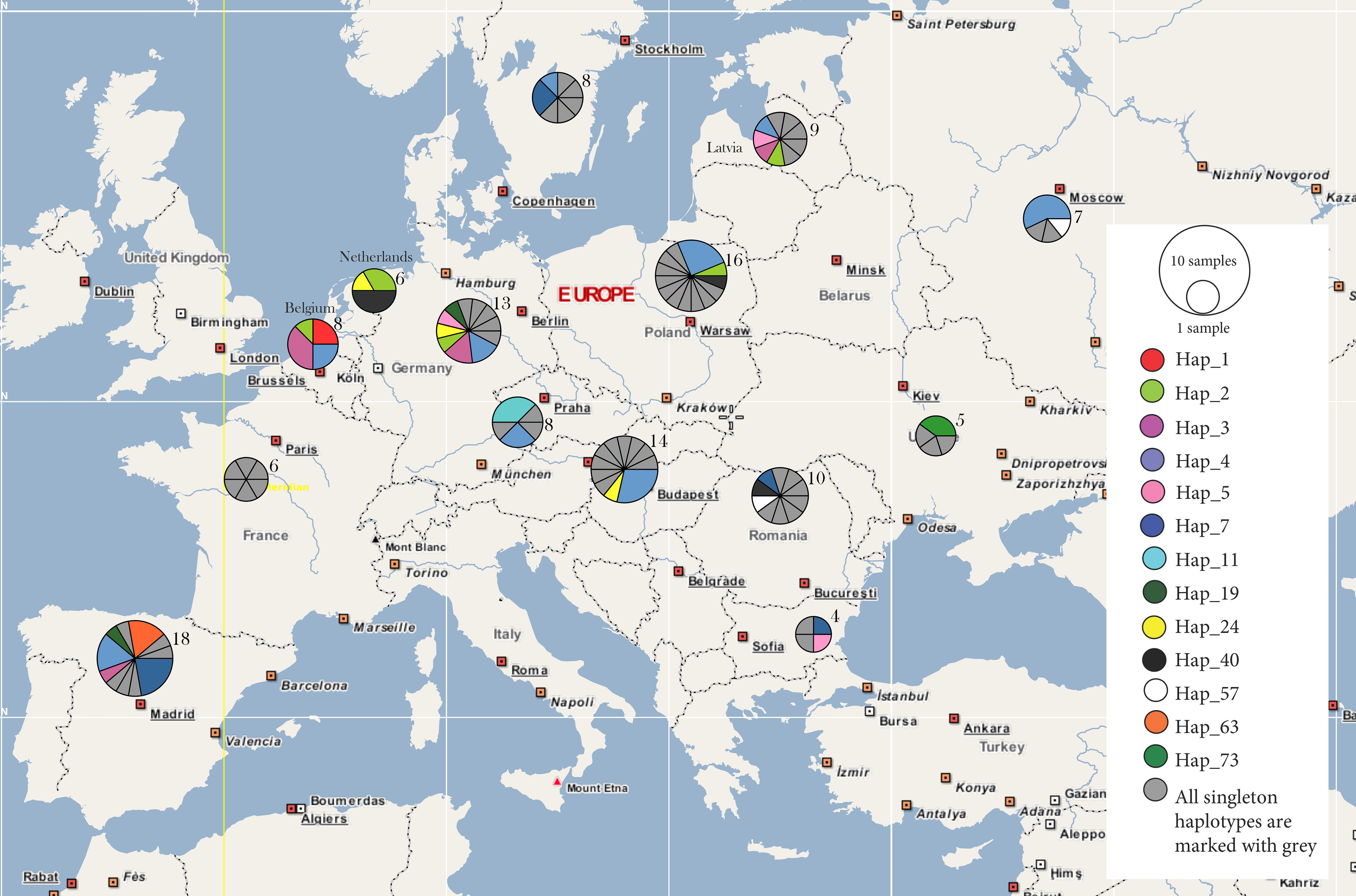
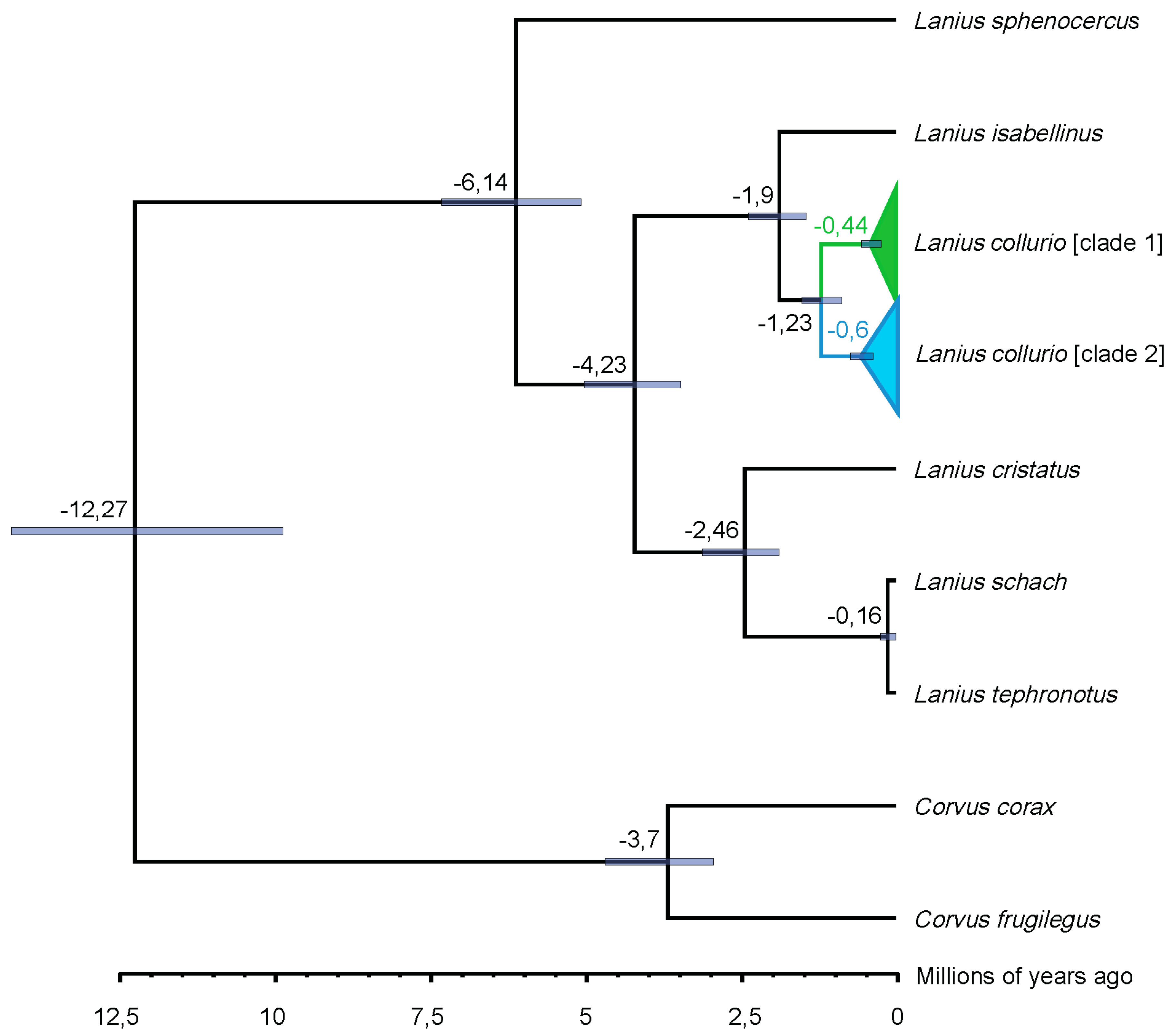
3. Results
4. Discussion
4.1. Behind the Mask: Genetic Panmixia
4.2. A Blast from the Past: Pleistocene Upbringing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

References
- Hewitt, G. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.R.; Lister, A.M.; Barnes, I.; Dalén, L. Refugia revisited: Individualistic responses of species in space and time. Proc. R. Soc. B Biol. Sci. 2010, 277, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Huybers, P. Glacial variability over the last two million years: An extended depth-derived agemodel, continuous obliquity pacing, and the Pleistocene progression. Quat. Sci. Rev. 2007, 26, 37–55. [Google Scholar] [CrossRef]
- Lisiecki, L.E.; Raymo, M.E. Plio-Pleistocene climate evolution: Trends and transitions in glacial cycle dynamics. Quat. Sci. Rev. 2007, 26, 56–69. [Google Scholar] [CrossRef]
- Pisias, N.G.; Moore, T.C. The evolution of Pleistocene climate: A time series approach. Earth Planet. Sci. Lett. 1981, 52, 450–458. [Google Scholar] [CrossRef]
- Hewitt, G.M. Speciation, hybrid zones and phylogeography—Or seeing genes in space and time. Mol. Ecol. 2001, 10, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Médail, F.; Diadema, K. Glacial refugia influence plant diversity patterns in the Mediterranean Basin. J. Biogeogr. 2009, 36, 1333–1345. [Google Scholar] [CrossRef]
- Hewitt, G.M. Post-glacial re-colonization of European biota. Biol. J. Linn. Soc. 1999, 68, 87–112. [Google Scholar] [CrossRef]
- Černa Bolfíková, B.; Eliášová, K.; Loudová, M.; Kryštufek, B.; Lymberakis, P.; Sándor, A.D.; Hulva, P. Glacial allopatry vs. postglacial parapatry and peripatry: The case of hedgehogs. PeerJ 2017, 5, e3163. [Google Scholar] [CrossRef] [PubMed]
- Webb, T., III. Glacial and Holocene vegetation history: Eastern North America. In Handbook of Vegetation Science; Huntley, B., Webb, T., III, Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1988; pp. 385–414. [Google Scholar]
- Taberlet, P.; Fumagalli, L.; Wust-Saucy, A.G.; Cosson, J.F. Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol. 1998, 7, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Remington, C.L. Suture-Zones of Hybrid Interaction Between Recently Joined Biotas. In Evolutionary Biology: Volume 2; Dobzhansky, T., Hecht, M.K., Steere, W.C., Eds.; Springer US: Boston, MA, USA, 1968; pp. 321–428. ISBN 978-1-4684-8094-8. [Google Scholar]
- Weiss, S.; Ferrand, N. Phylogeography of Southern European Refugia; Springer: Dordrecht, The Netherlands, 2007; ISBN 978-1-4020-4903-3. [Google Scholar]
- Avise, J.C. Evolutionary Pathways in Nature: A Phylogenetic Approach; Cambridge University Press: Cambridge, UK, 2006; ISBN 9780511606939. [Google Scholar]
- Wiley, E.O.; Lieberman, B.S. Phylogenetics: Theory and Practice of Phylogenetic Systematics, 2nd ed.; Willey Blackwell: Hoboken, NJ, USA, 2011; ISBN 9780470905968. [Google Scholar]
- Del Hoyo, J.; Collar, N.J. Illustrated Checklist of the Birds of the World, Volume 2: Passerines; Lynx Edicions: Barcelona, Spain, 2016; ISBN 978-84-96553-98-9. [Google Scholar]
- Gill, F.; Donsker, D. IOC World Bird List v8.1. Available online: http://www.worldbirdnames.org/ (accessed on 16 February 2018).
- Kvist, L.; Giralt, D.; Valera, F.; Hoi, H.; Kristin, A.; Darchiashvili, G.; Lovaszi, P. Population decline is accompanied by loss of genetic diversity in the Lesser Grey Shrike Lanius minor. Ibis (Lond. 1859) 2011, 153, 98–109. [Google Scholar] [CrossRef]
- Olsson, U.; Alström, P.; Svensson, L.; Aliabadian, M.; Sundberg, P. The Lanius excubitor (Aves, Passeriformes) conundrum-Taxonomic dilemma when molecular and non-molecular data tell different stories. Mol. Phylogenet. Evol. 2010, 55, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Padilla, D.P.; Spurgin, L.G.; Fairfield, E.A.; Illera, J.C.; Richardson, D.S. Population history, gene flow, and bottlenecks in island populations of a secondary seed disperser, the southern grey shrike (Lanius meridionalis koenigi). Ecol. Evol. 2015, 5, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.; Wink, M.; Garcia-del-Rey, E.; Delgado Castro, G. Evidence from DNA nucleotide sequences and ISSR profiles indicates paraphyly in subspecies of the Southern Grey Shrike (Lanius meridionalis). J. Ornithol. 2008, 149, 495–506. [Google Scholar] [CrossRef]
- Mundy, N.I.; Winchell, C.S.; Woodruff, D.S. Genetic differences between the endangered San Clemente Island loggerhead shrike Lanius ludovicianus mearnsi and two neighbouring subspecies demonstrated by mtDNA control region and cytochrome b sequence variation. Mol. Ecol. 1997, 6, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Caballero, I.C.; Ashley, M.V. Genetic analysis of the endemic island loggerhead shrike, Lanius ludovicianus anthonyi. Conserv. Genet. 2011, 12, 1485–1493. [Google Scholar] [CrossRef]
- del Hoyo, J.; Elliott, A.; Christie, D.A. (Eds.) Handbook of the Birds of the World. Vol. 13. Penduline-Tits to Shrikes; Lynx Edicions: Barcelona, Spain, 2008. [Google Scholar]
- Euro Bird Portal. Available online: http://www.eurobirdportal.org/ebp/en/#home/LANCOL/r2000/LANTOR/r2000/ (accessed on 1 March 2018).
- IUCN Red List. Available online: https://www.iucnredlist.org/species/22705001/110988087 (accessed on 31 October 2018).
- Brochet, A.-L.; Van den Bosschen, W.; Jbour, S.; Ndang’ang’a, P.K.; Jones, V.R.; Abdou, W.A.L.I.; Al-hmoud, A.R.; Asswad, N.G.; Atienza, J.C.; Atrash, I.; et al. Preliminary assessment of the scope and scale of illegal killing and taking of birds in the Mediterranean. Bird Conserv. Int. 2016, 26, 1–28. [Google Scholar] [CrossRef]
- Eason, P.; Rabia, B.; Attum, O. Hunting of migratory birds in North Sinai, Egypt. Bird Conserv. Int. 2016, 26, 39–51. [Google Scholar] [CrossRef]
- Allendorf, F.W.; Luikart, G.; Aitken, S.N. Conservation and the Genetics of Populations, 2nd ed.; Willey Blackwell: Hoboken, NJ, USA, 2012. [Google Scholar]
- Hartl, D.L.; Clark, A.G. Principles of Population Genetics, 3rd ed.; Sinauer Associates, Inc.: Sunderland, MA, USA, 1997; ISBN 0-87893-308-5. [Google Scholar]
- Krebs, J.E.; Goldstein, E.S.; Kilpatrick, S.T. Lewin’s Genes XII; Jones & Bartlett Learning: Burlington, MA, USA, 2017; ISBN 9780763766320. [Google Scholar]
- Mindell, D.P. (Ed.) Avian Molecular Evolution and Systematics; Academic Press: London, UK, 1997. [Google Scholar]
- Rubinoff, D.; Holland, B.S. Between two extremes: Mitochondrial DNA is neither the panacea nor the nemesis of phylogenetic and taxonomic inference. Syst. Biol. 2005, 54, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Hurst, G.D.D.; Jiggins, F.M. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: The effects of inherited symbionts. Proc. R. Soc. B Biol. Sci. 2005, 272, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, F.; Alexandrino, J.; Weiss, S.; Ferrand, N. Documenting the advantages and limitations of different classes of molecular markers in a well-established phylogeographic context: Lessons from the Iberian endemic Golden-striped salamander, Chioglossa lusitanica (Caudata: Salamandridae). Biol. J. Linn. Soc. 2008, 95, 371–387. [Google Scholar] [CrossRef]
- Green, M.R.; Sambrook, J. Molecular Cloning: A Laboratory Manual, Vol. 1, 4th ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2012. [Google Scholar]
- Sorenson, M.D.; Ast, J.C.; Dimcheff, D.E.; Yuri, T.; Mindell, D.P. Primers for a PCR-based approach to mitochondrial genome sequencing in birds and other vertebrates. Mol. Phylogenet. Evol. 1999, 12, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, M.D.; Quinn, T.W. Numts: A Challenge for Avian Systematics and Population Biology. Auk 1998, 115, 214–221. [Google Scholar] [CrossRef]
- Miranda, H.C.; Kennedy, R.S.; Mindell, D.P. Phylogenetic placement of Mimizuku gurneyi (Aves: Strigidae) inferred from mitochondrial DNA. Auk 1997, 114, 315–323. [Google Scholar] [CrossRef]
- Sorenson, M.D. Avian mtDNA Primers; Boston University: Boston, MA, USA, 2003. [Google Scholar]
- Wink, M.; Sauer-Gürth, H.; Heidrich, P.; Witt, H.-H.; Gwinner, E. A Molecular Phylogeny of Stonechats and Related Turdids. In A Guide to the Genus Saxicola; Christopher Helm: London, UK, 2002; pp. 22–30. [Google Scholar]
- Johnsen, A.; Rindal, E.; Ericson, P.G.P.; Zuccon, D.; Kerr, K.C.R.; Stoeckle, M.Y.; Lifjeld, J.T. DNA barcoding of Scandinavian birds reveals divergent lineages in trans-Atlantic species. J. Ornithol. 2010, 151, 565–578. [Google Scholar] [CrossRef]
- Lohman, D.J.; Prawiradilaga, D.M.; Meier, R. Improved COI barcoding primers for Southeast Asian perching birds (Aves: Passeriformes). Mol. Ecol. Resour. 2009, 9, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Guindon, S.; Gascuel, O. Sea view version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar] [CrossRef]
- Weir, J.T.; Schluter, D. Calibrating the avian molecular clock. Mol. Ecol. 2008, 17, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A. FigTree v1. 3.1; Institute of Evolutionary Biology, University of Edinburgh: Edinburgh, UK, 2010. [Google Scholar]
- Wang, E.; van Wijk, R.E.; Braun, M.S.; Wink, M. Gene flow and genetic drift contribute to high genetic diversity with low phylogeographical structure in European Hoopoes (Upupa epops). Mol. Phylogenet. Evol. 2017, 113, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Resano-Mayor, J.; Fernández-Martín, Á.; Hernández-Gómez, S.; Toranzo, I.; España, A.; Gil, J.A.; de Gabriel, M.; Roa-Álvarez, I.; Strinella, E.; Hobson, K.A.; et al. Integrating genetic and stable isotope analyses to infer the population structure of the White-winged Snowfinch Montifringilla nivalis in Western Europe. J. Ornithol. 2017, 158, 395–405. [Google Scholar] [CrossRef]
- Zink, R.M.; Pavlova, A.; Drovetski, S.; Rohwer, S. Mitochondrial phylogeographies of five widespread Eurasian bird species. J. Ornithol. 2008, 149, 399–413. [Google Scholar] [CrossRef]
- Arbabi, T.; Gonzalez, J.; Wink, M. Mitochondrial evidence for genetic diversity and low phylogeographic differentiation in the Marsh Warbler Acrocephalus palustris (Aves: Acrocephalidae). Org. Divers. Evol. 2014, 14, 409–417. [Google Scholar] [CrossRef]
- Zehtindjiev, P.; Ilieva, M.; Hansson, B.; Oparina, O.; Oparin, M.; Bensch, S. Population genetic structure in the paddyfield warbler (Acrocephalus agricola Jerd.). Curr. Zool. 2011, 57, 63–71. [Google Scholar] [CrossRef]
- Arbabi, T.; Gonzalez, J.; Witt, H.H.; Klein, R.; Wink, M. Mitochondrial phylogeography of the Eurasian Reed Warbler Acrocephalus scirpaceus and the first genetic record of A. s. fuscus in Central Europe. Ibis (Lond. 1859) 2014, 156, 799–811. [Google Scholar] [CrossRef]
- Rutkowski, R.; Zawadzka, D.; Merta, D.; Stanković, A.; Jagołkowska, P.; Suchecka, E.; Kobielski, J. Conservation genetics of the Capercaillie Tetrao urogallus in Poland—Diversity of mitochondrial DNA in remnant and extinct populations. Acta Ornithol. 2017, 52, 179–196. [Google Scholar] [CrossRef]
- Rutkowski, R.; Jagołkowska, P.; Zawadzka, D.; Bogdanowicz, W. Impacts of forest fragmentation and post-glacial colonization on the distribution of genetic diversity in the Polish population of the hazel grouse Terastes bonasia. Eur. J. Wildl. Res. 2016, 62, 293–306. [Google Scholar] [CrossRef]
- Perez-Tris, J.; Bensch, S.; Carbonell, R.; Helbig, A.J.; Telleria, J.L. Historical diversification of migration patterns in a passerine bird. Evolution (N. Y.) 2004, 58, 1819–1832. [Google Scholar] [CrossRef]
- Carneiro de Melo Moura, C.; Bastian, H.-V.; Bastian, A.; Wang, E.; Wang, X.; Wink, M. Pliocene Origin, Ice Ages and Postglacial Population Expansion Have Influenced a Panmictic Phylogeography of the European Bee-Eater. Diversity 2019, 11, 12. [Google Scholar] [CrossRef]
- Aoki, D.; Kinoshita, G.; Kryukov, A.P.; Nishiumi, I.; Lee, S.; Suzuki, H. Quaternary-related genetic differentiation and parallel population dynamics of the Eurasian Jay (Garrulus glandarius) in the circum-Japan Sea region. J. Ornithol. 2018, 159, 1087–1097. [Google Scholar] [CrossRef]
- Cramp, S.; Perrins, C.M. (Eds.) The Birds of the Western Palearctic; Oxford University Press: Oxford, UK, 1993. [Google Scholar]
- Tryjanowski, P.; Goławski, A.; Kuźniak, S.; Mokwa, T.; Antczak, M. Disperse or stay? Exceptionally high breeding-site infidelity in the red-backed shrike Lanius collurio. Ardea 2007, 95, 316–320. [Google Scholar] [CrossRef]
- Giralt, D. Decline of the Lesser Grey Shrike (Lanius minor) at the Western Limit of the Distribution Area: Causes, Mechanisms and Conservation Proposals; University of Barcelona: Barcelona, Spain, 2015. [Google Scholar]
- Fuchs, J.; Crowe, T.M.; Bowie, R.C.K. Phylogeography of the fiscal shrike (Lanius collaris): A novel pattern of genetic structure across the arid zones and savannas of Africa. J. Biogeogr. 2011, 38, 2210–2222. [Google Scholar] [CrossRef]
- Sittenthaler, M.; Kunz, F.; Szymusik, A.; Grünschachner-Berger, V.; Krumböck, S.; Stauffer, C.; Nopp-Mayr, U. Fine-scale genetic structure in an eastern Alpine black grouse Tetrao tetrix metapopulation. J. Avian Biol. 2018, 49, 1–14. [Google Scholar] [CrossRef]
- Hogner, S.; Laskemoen, T.; Lifjeld, J.T.; Porkert, J.; Kleven, O.; Albayrak, T.; Kabasakal, B.; Johnsen, A. Deep sympatric mitochondrial divergence without reproductive isolation in the common redstart Phoenicurus phoenicurus. Ecol. Evol. 2012, 2, 2974–2988. [Google Scholar] [CrossRef] [PubMed]
- Bagi, Z.; Dimopoulos, E.A.; Loukovitis, D.; Eraud, C.; Kusza, S. MtDNA genetic diversity and structure of Eurasian Collared Dove (Streptopelia decaocto). PLoS ONE 2018, 13, e0193935. [Google Scholar] [CrossRef] [PubMed]
- Kamp, L.; Pasinelli, G.; Milanesi, P.; Drovetski, S.V.; Kosiński, Z.; Kossenko, S.; Robles, H.; Schweizer, M. Significant Asia-Europe divergence in the middle spotted woodpecker (Aves, Picidae). Zool. Scr. 2018, 1–16. [Google Scholar] [CrossRef]
- Pellegrino, I.; Negri, A.; Cucco, M.; Mucci, N.; Pavia, M.; Šálek, M.; Boano, G.; Randi, E. Phylogeography and Pleistocene refugia of the Little Owl Athene noctua inferred from mtDNA sequence data. Ibis (Lond. 1859) 2014, 156, 639–657. [Google Scholar] [CrossRef]
- Hansson, B.; Hasselquist, D.; Tarka, M.; Zehtindjiev, P.; Bensch, S. Postglacial colonisation patterns and the role of isolation and expansion in driving diversification in a passerine bird. PLoS ONE 2008, 3, e2794. [Google Scholar] [CrossRef] [PubMed]
- Brito, P.H. The influence of Pleistocene glacial refugia on tawny owl genetic diversity and phylogeography in Western Europe. Mol. Ecol. 2005, 14, 3077–3094. [Google Scholar] [CrossRef] [PubMed]
- Webb, W.C.; Marzluff, J.M.; Omland, K.E. Random interbreeding between cryptic lineages of the Common Raven: Evidence for speciation in reverse. Mol. Ecol. 2011, 20, 2390–2402. [Google Scholar] [CrossRef] [PubMed]
- Quinn, T.W. The genetic legacy of Mother Goose—Phylogeographic patterns of lesser snow goose Chen caerulescens caerulescens maternal lineages. Mol. Ecol. 1992, 1, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Tottrup, A.P.; Klaassen, R.H.G.; Strandberg, R.; Thorup, K.; Kristensen, M.W.; Jorgensen, P.S.; Fox, J.; Afanasyev, V.; Rahbek, C.; Alerstam, T. The annual cycle of a trans-equatorial Eurasian-African passerine migrant: Different spatio-temporal strategies for autumn and spring migration. Proc. R. Soc. B Biol. Sci. 2012, 279, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.; Fraser, K.C.; Kyser, T.K.; Tøttrup, A.P. Combining direct and indirect tracking techniques to assess the impact of sub-Saharan conditions on cross-continental songbird migration. J. Ornithol. 2016, 157, 1037–1047. [Google Scholar] [CrossRef]
- Tøttrup, A.P.; Pedersen, L.; Onrubia, A.; Klaassen, R.H.G.; Thorup, K. SM Migration of red-backed shrikes from the Iberian Peninsula: Optimal or sub-optimal detour? J. Avian Biol. 2017, 48, 149–154. [Google Scholar] [CrossRef]
- Titeux, N.; Van der Elst, D.; Van Nieuwenhuyse, D. Pie-grièche écorcheur, Lanius collurio. In Atlas Des Oiseaux Nicheurs de Wallonie 2001–2007; Jacob, J.-P., Dehem, C., Burnel, A., Dambiermont, J.-L., Fasol, M., Kinet, T., Van der Elst, D., Paquet, J.-Y., Eds.; Aves & Région Wallonne: Gembloux, Belgium, 2010; pp. 396–397. [Google Scholar]
- Derouaux, A.; Paquet, J.-Y. The worrying trends in breeding bird populations of Wallonia: 28 years of common bird monitoring. Bull. Aves 2018, 55, 1–31. [Google Scholar]
- Tellería, J.L. Distribution of the Red-Backed Shrike Lanius Collurio at Its Western Range Boundary: Patterns and Conservation Prospects. Ardeola 2018, 65, 221–232. [Google Scholar] [CrossRef]
- Tellería, J.L. Old Counts Suggest the Collapse of Two Red-Backed Shrike Lanius collurio Populations. Ardeola 2018, 65, 283–290. [Google Scholar] [CrossRef]
- Birdlife International. The Killing; Birdlife International: Cambridge, UK, 2015. [Google Scholar]
- Birdlife International. The Killing 2.0. A view to Kill; Birdlife International: Cambridge, UK, 2017. [Google Scholar]
- Hirschfeld, A.; Attard, G. Vogeljagd in Europa—Analyse von abschusszahlen und auswirkungen der jagd auf den erhalt bedrohter Vogelarten. Vogelschutz 2017, 53, 15–42. [Google Scholar]
- Hewitt, G.M.; Butlin, R.K. Causes and consequences of population structure. In Behavioural Ecology: An Evolutionary Approach; Krebs, J.R., Davies, N.B., Eds.; Blackwell: Oxford, UK, 1997; pp. 350–372. [Google Scholar]
- King, R.A.; Ferris, C. Chloroplast DNA phylogeography of Alnus glutinosa (L.) Gaertn. Mol. Ecol. 1998, 7, 1151–1161. [Google Scholar] [CrossRef]
- Brewer, S.; Cheddadi, R.; de Beaulieu, J.L.; Reille, M. The spread of deciduous Quercus throughout Europe since the last glacial period. For. Ecol. Manag. 2002, 156, 27–48. [Google Scholar] [CrossRef]
- Schrimpf, A.; Theissinger, K.; Dahlem, J.; Maguire, I.; Pârvulescu, L.; Schulz, H.K.; Schulz, R. Phylogeography of noble crayfish (Astacus astacus) reveals multiple refugia. Freshw. Biol. 2014, 59, 761–776. [Google Scholar] [CrossRef]
- Fijarczyk, A.; Nadachowska, K.; Hofman, S.; Litvinchuk, S.N.; Babik, W.; Stuglik, M.; Gollmann, G.; Choleva, L.; Cogǎlniceanu, D.; Vukov, T.; et al. Nuclear and mitochondrial phylogeography of the European fire-bellied toads Bombina bombina and Bombina variegata supports their independent histories. Mol. Ecol. 2011, 20, 3381–3398. [Google Scholar] [CrossRef] [PubMed]
- Lenk, P.; Fritz, U.; Joger, U.; Wink, M. Mitochondrial phylogeography of the European pond turtle, Emys orbicularis (Linnaeus 1758). Mol. Ecol. 1999, 8, 1911–1922. [Google Scholar] [CrossRef] [PubMed]
- Mezzasalma, M.; Di Febbraro, M.; Guarino, F.M.; Odierna, G.; Russo, D. Cold-blooded in the Ice Age: “refugia within refugia”, inter-and intraspecific biogeographic diversification of European whipsnakes (Squamata, Colubridae, Hierophis). Zoology 2018. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.T.; Bellaagh, M.; Wink, M.; Paunović, A.; Korsós, Z. Phylogeography of the Caspian whipsnake in Europe with emphasis on the westernmost populations. Amphib. Reptil. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Psonis, N.; Antoniou, A.; Karameta, E.; Leaché, A.D.; Kotsakiozi, P.; Darriba, D.; Kozlov, A.; Stamatakis, A.; Poursanidis, D.; Kukushkin, O.; et al. Resolving complex phylogeographic patterns in the Balkan Peninsula using closely related wall-lizard species as a model system. Mol. Phylogenet. Evol. 2018, 125, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Kohn, M.; Knauer, F.; Stoffella, A.; Schröder, W.; Pääbo, S. Conservation genetics of the European brown bear—A study using excremental PCR of nuclear and mitochondrial sequences. Mol. Ecol. 1995, 4, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Dietzen, C.; Garcia-del-Rey, E.; Castro, G.D.; Wink, M. Phylogeography of the blue tit (Parus teneriffae-group) on the Canary Islands based on mitochondrial DNA sequence data and morphometrics. J. Ornithol. 2008, 149, 1–12. [Google Scholar] [CrossRef]
- Carneiro de Melo Moura, C.; de Araujo, H.F.; Aleixo, A.; Wink, M.; Fernandes, A.M. The role of landscape change and paleoclimatic events in shaping the evolutionary history of the Polioptila gnatcatchers (Passeriformes, Polioptilidae) with emphasis on species associated with open habitats. J. Avian Biol. 2018, 49, 1–12. [Google Scholar] [CrossRef]
- Kraus, R.H.S.; Wink, M. Avian genomics: Fledging into the wild! J. Ornithol. 2015, 156, 851–865. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | π | Hd | Ss | Nh | N |
|---|---|---|---|---|---|
| Belgium | 0.013 | 0.821 | 36 | 4 | 8 |
| Bulgaria | 0.002 | 1 | 7 | 4 | 4 |
| Czech Republic | 0.004 | 0.857 | 22 | 5 | 8 |
| France | 0.014 | 1 | 42 | 6 | 6 |
| Germany | 0.012 | 0.974 | 43 | 11 | 13 |
| Hungary | 0.009 | 0.934 | 49 | 11 | 14 |
| Latvia | 0.009 | 1 | 42 | 9 | 9 |
| Netherlands | 0.012 | 0.733 | 33 | 3 | 6 |
| Poland | 0.011 | 0.95 | 52 | 13 | 16 |
| Romania | 0.006 | 1 | 41 | 10 | 10 |
| Russia | 0.001 | 0.714 | 7 | 4 | 7 |
| Spain | 0.003 | 0.921 | 41 | 11 | 18 |
| Sweden | 0.013 | 0.964 | 42 | 7 | 8 |
| Ukraine | 0.002 | 0.9 | 8 | 4 | 5 |
| Average/Total | 0.009 | 0.96 | 110 | 76 | 132 |
| Belgium | Bulgaria | CzechRep | France | Germany | Hungary | Latvia | Netherlands | Poland | Romania | Russia | Spain | Sweden | Ukraine | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Belgium | * | + | + | |||||||||||
| Bulgaria | 0.1040 | * | ||||||||||||
| CzechRep | 0.1047 | 0.0828 | * | + | + | + | ||||||||
| France | 0.0943 | 0 | 0.0753 | * | + | |||||||||
| Germany | −0.0082 | −0.0041 | 0.0440 | 0.0141 | * | + | ||||||||
| Hungary | 0.0505 | 0.0397 | 0.0328 | 0.0367 | −0.0037 | * | + | |||||||
| Latvia | 0.0050 | −0.0285 | 0.0436 | 0.0000 | −0.0401 | 0.0027 | * | |||||||
| Netherlands | 0.1857 | 0.1489 | 0.2005 | 0.1333 | 0.0963 | 0.1411 | 0.0896 | * | + | + | + | + | ||
| Poland | 0.0419 | 0.0303 | 0.0325 | 0.0280 | −0.0055 | −0.0146 | −0.0088 | 0.0953 | * | |||||
| Romania | 0.0861 | −0.0256 | 0.0691 | 0.0000 | 0.0132 | 0.0341 | 0.0000 | 0.0754 | 0.0200 | * | + | |||
| Russia | 0.1025 | 0.1649 | 0.0813 | 0.1479 | 0.0620 | 0.0049 | 0.0783 | 0.2766 | 0.0168 | 0.1211 | * | + | ||
| Spain | 0.0653 | −0.0090 | 0.0688 | 0.0445 | 0.0148 | 0.0259 | 0.0174 | 0.1553 | 0.0237 | 0.0194 | 0.0819 | * | + | |
| Sweden | 0.0783 | −0.0452 | 0.0599 | 0.0187 | 0.0114 | 0.0166 | 0.0038 | 0.1440 | 0.0125 | −0.0078 | 0.0928 | −0.0194 | * | |
| Ukraine | 0.1435 | 0.0530 | 0.1237 | 0.0480 | 0.0572 | 0.0801 | 0.0447 | 0.1876 | 0.0706 | 0.0440 | 0.2013 | 0.0871 | 0.0648 | * |
| Lc_1 | Lc_2 | Laniidae | Corvidae | |
|---|---|---|---|---|
| Lc_1 | * | |||
| Lc_2 | 0.022 | * | ||
| Laniidae | 0.068 | 0.071 | * | |
| Corvidae | 0.137 | 0.144 | 0.150 | * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pârâu, L.G.; Frias-Soler, R.C.; Wink, M. High Genetic Diversity among Breeding Red-Backed Shrikes Lanius collurio in the Western Palearctic. Diversity 2019, 11, 31. https://doi.org/10.3390/d11030031
Pârâu LG, Frias-Soler RC, Wink M. High Genetic Diversity among Breeding Red-Backed Shrikes Lanius collurio in the Western Palearctic. Diversity. 2019; 11(3):31. https://doi.org/10.3390/d11030031
Chicago/Turabian StylePârâu, Liviu G., Roberto Carlos Frias-Soler, and Michael Wink. 2019. "High Genetic Diversity among Breeding Red-Backed Shrikes Lanius collurio in the Western Palearctic" Diversity 11, no. 3: 31. https://doi.org/10.3390/d11030031
APA StylePârâu, L. G., Frias-Soler, R. C., & Wink, M. (2019). High Genetic Diversity among Breeding Red-Backed Shrikes Lanius collurio in the Western Palearctic. Diversity, 11(3), 31. https://doi.org/10.3390/d11030031