Impact of Land Use on Bacterial Diversity and Community Structure in Temperate Pine and Indigenous Forest Soils



Abstract
:1. Introduction
2. Materials and Methods
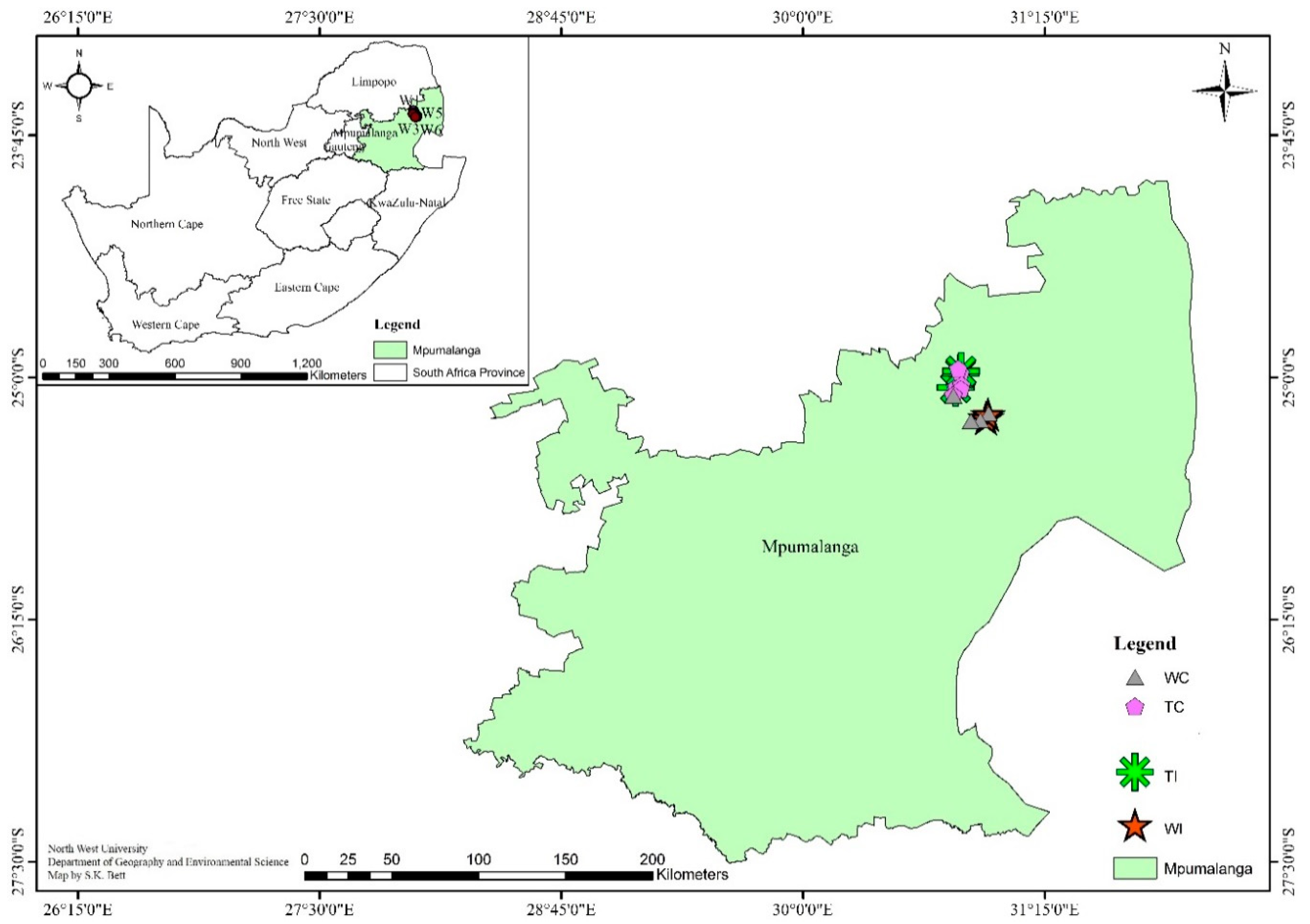
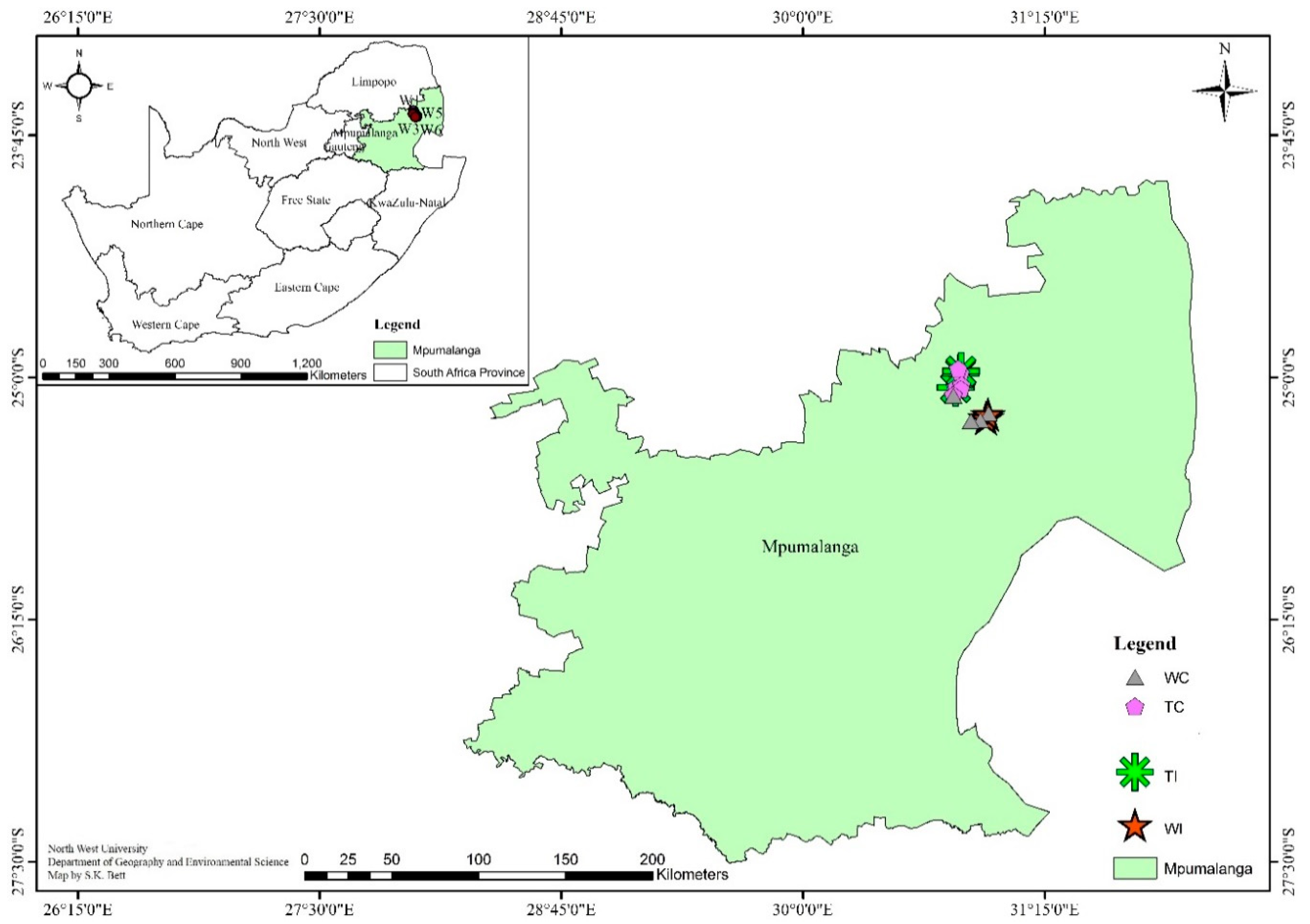
2.1. Site Description and Soil Sampling
2.2. Analyses of Soil Properties
2.3. Determination of Relative Bacterial Diversity and Taxonomic Richness
2.4. Statistical Analyses
3. Results
3.1. Soil Properties (Physical and Chemical) of the Forest Soil Samples
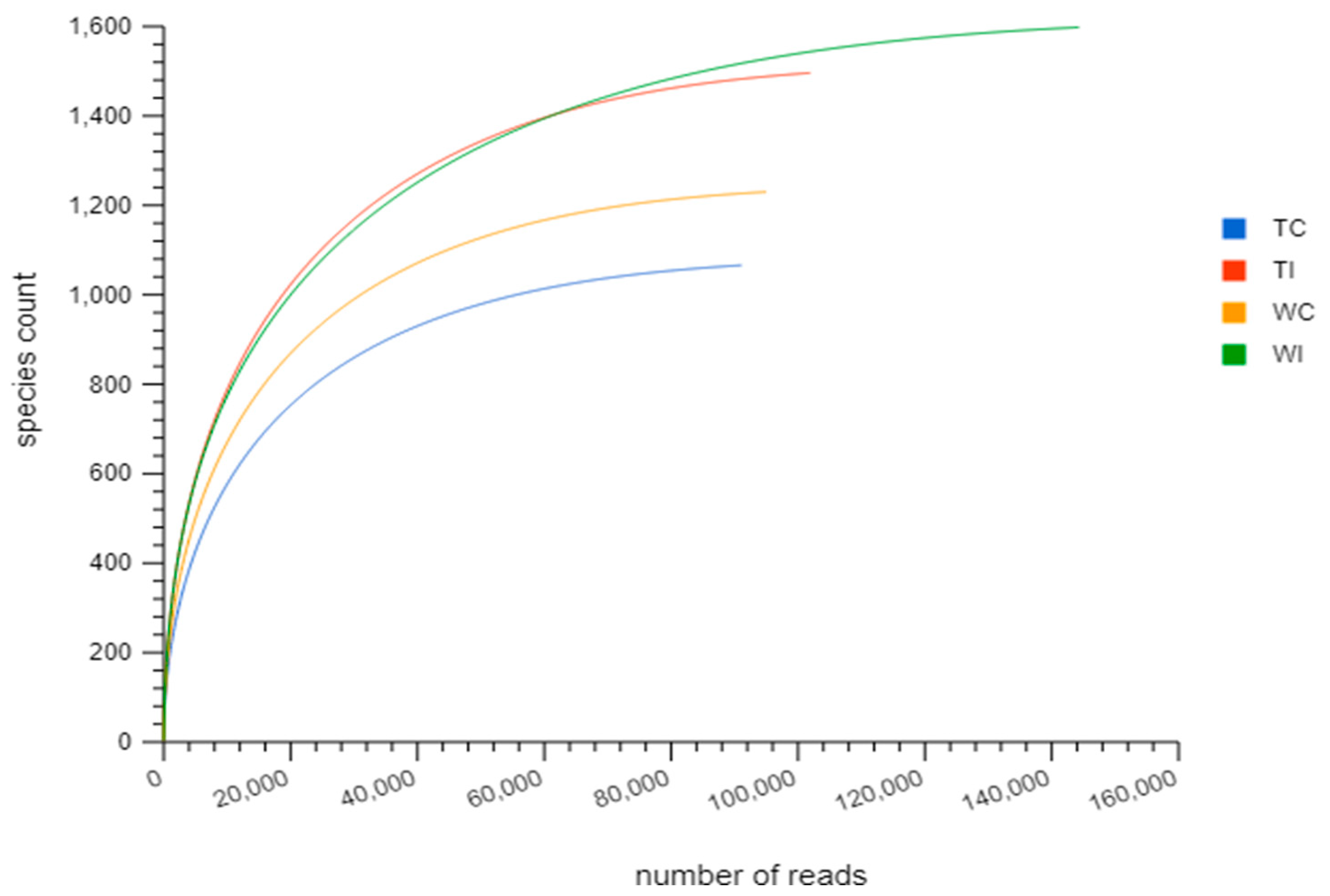
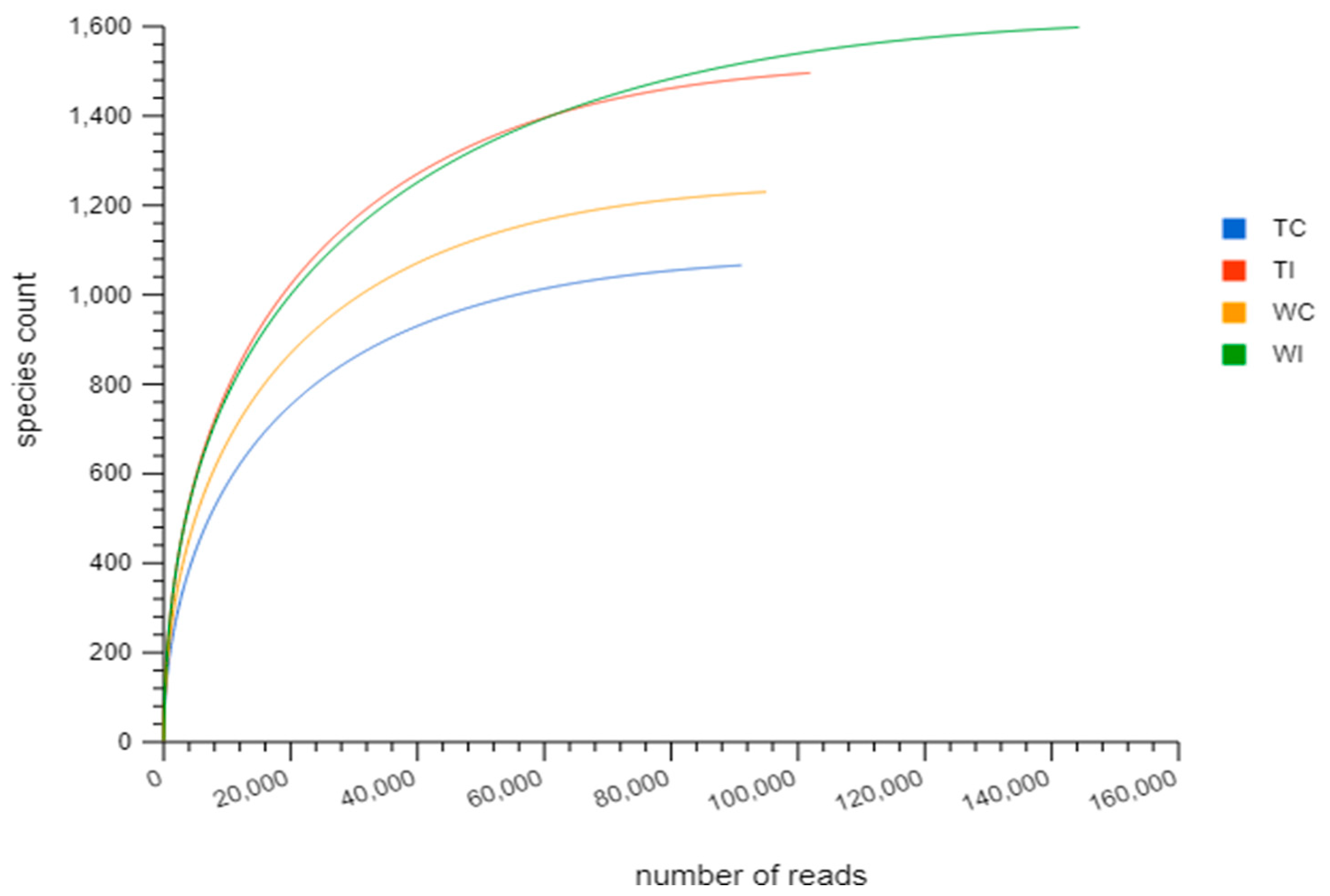
3.2. Rarefaction Analysis
3.3. Assessment of Diversity Indices
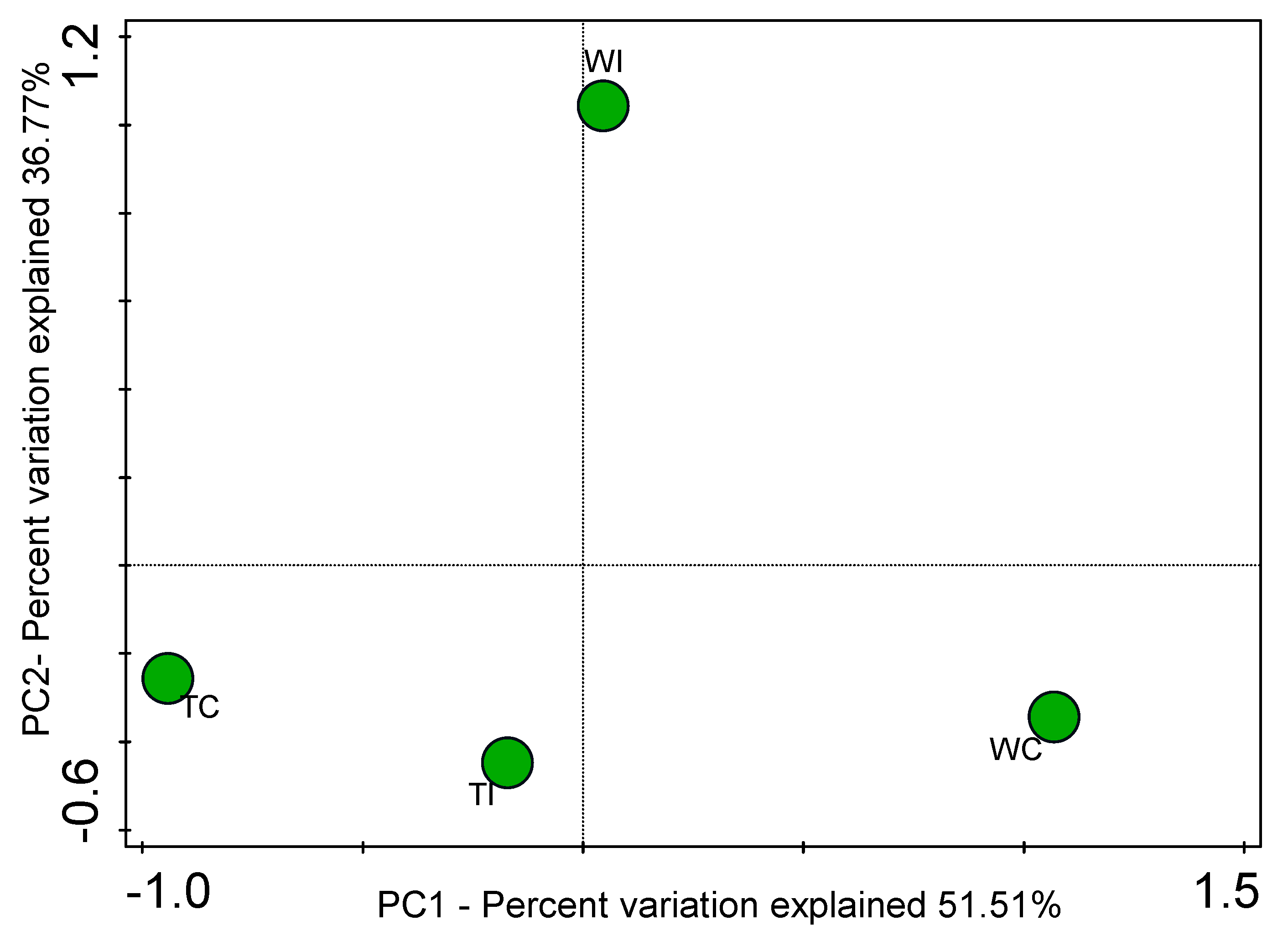
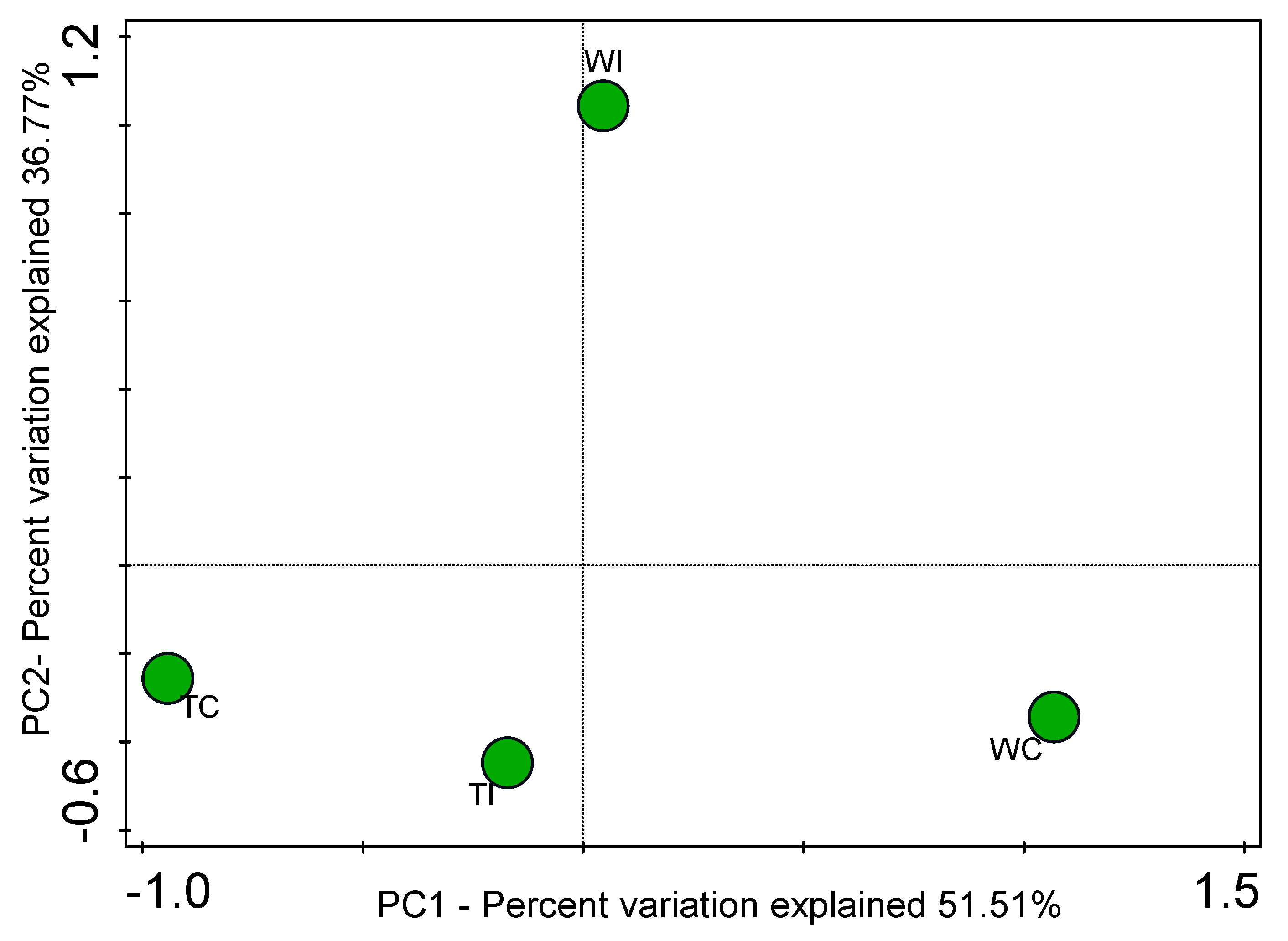
3.4. Principal Coordinates Analysis
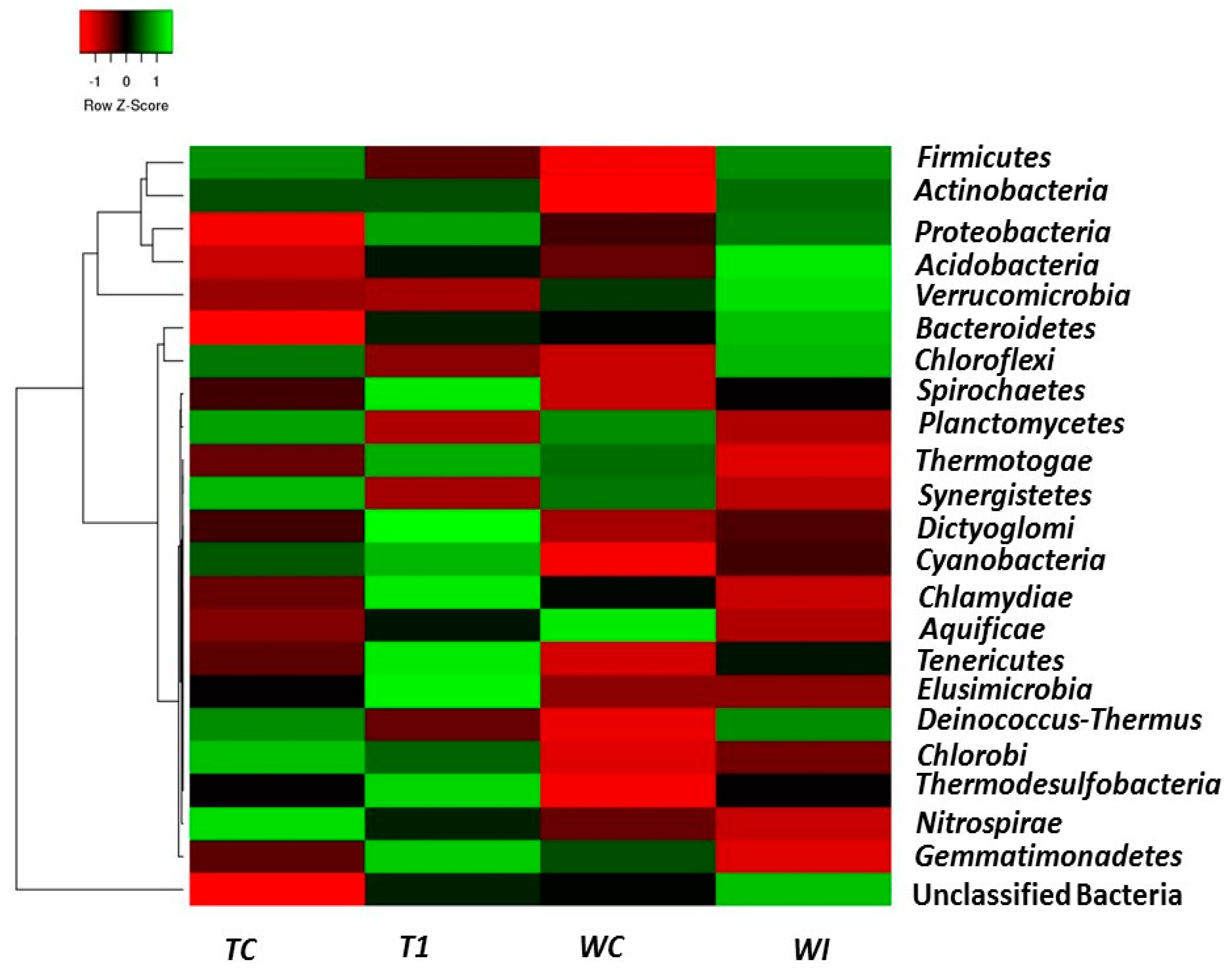
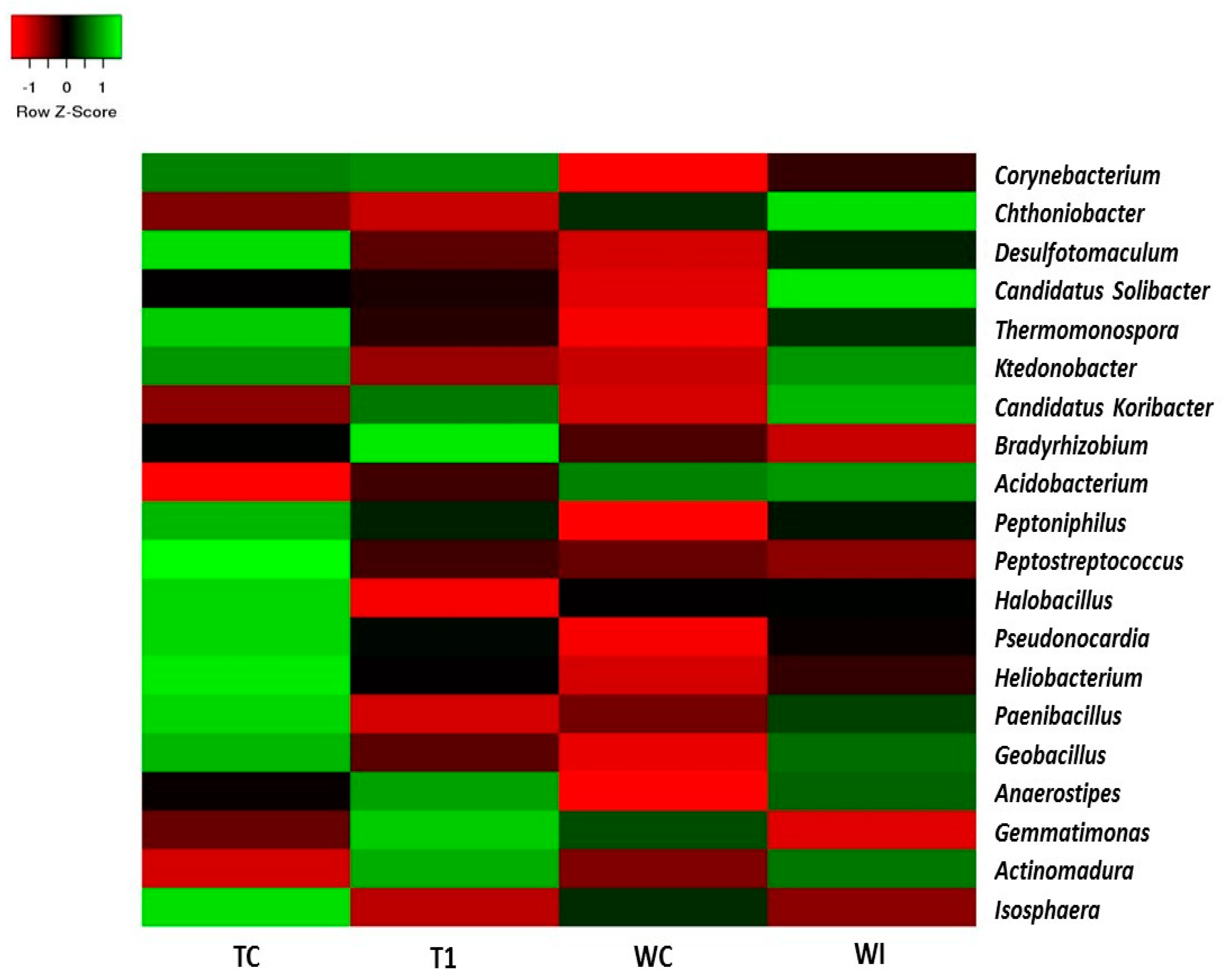
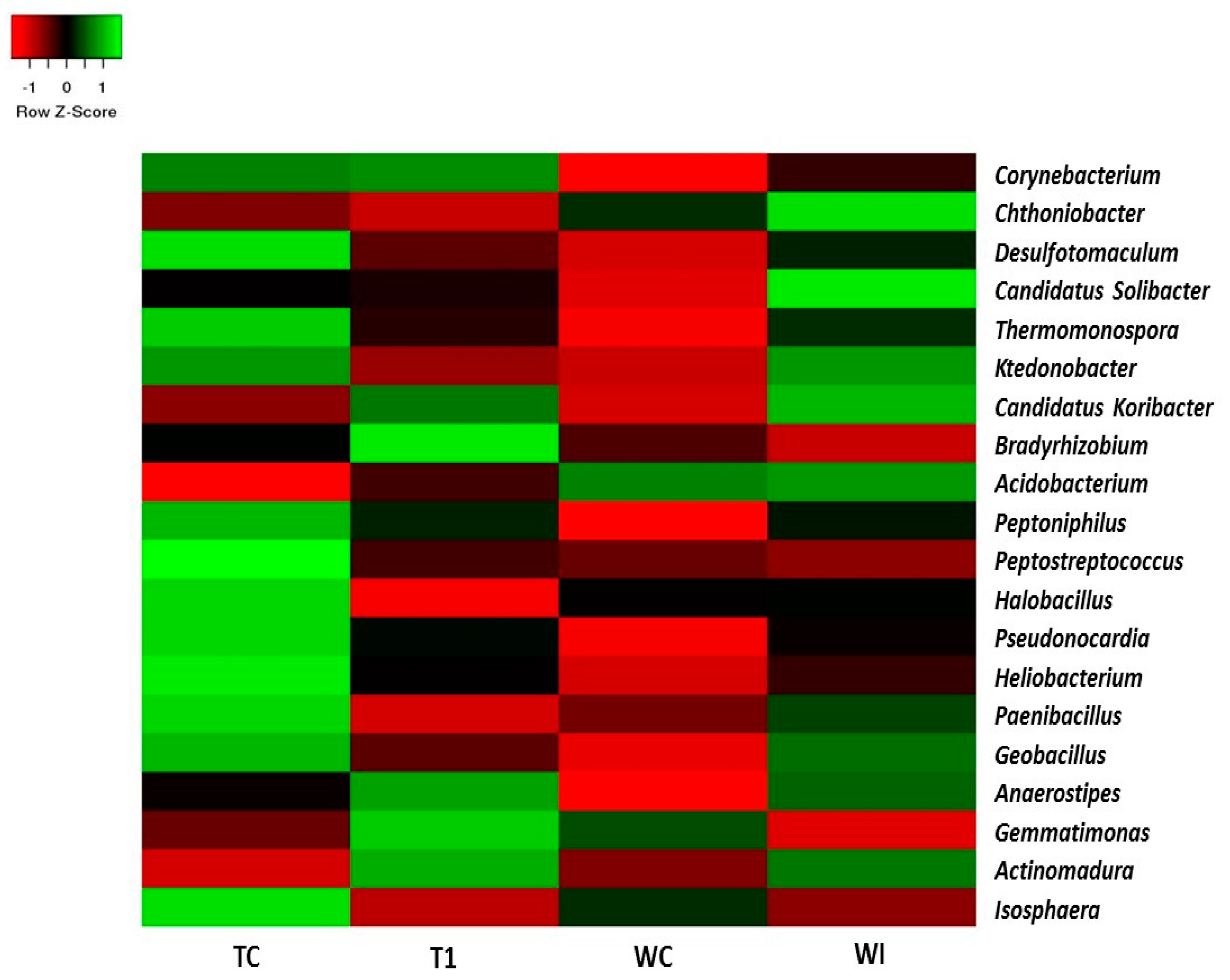
3.5. Phylum and Genus Level Distributions of the Bacterial Communities
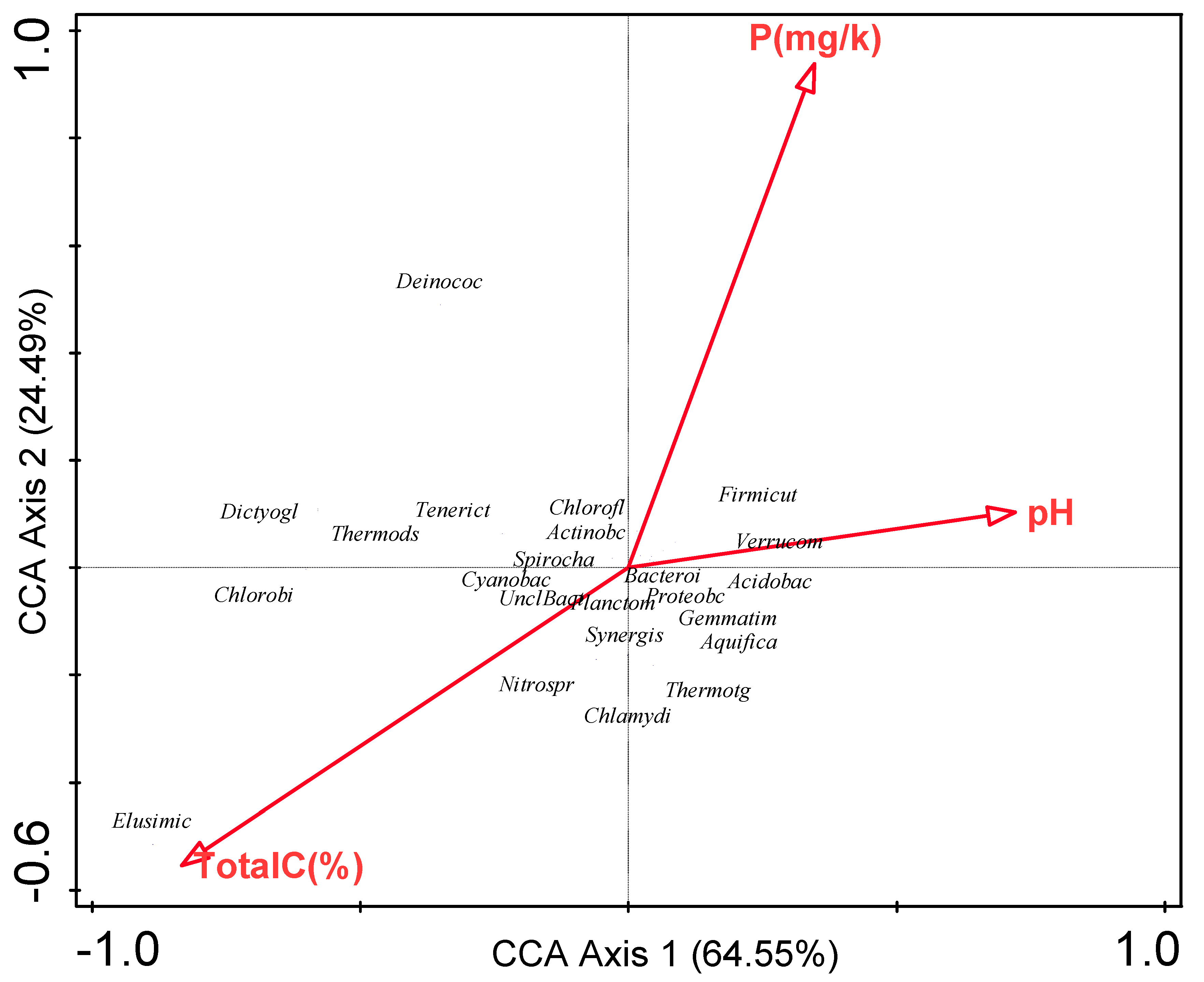
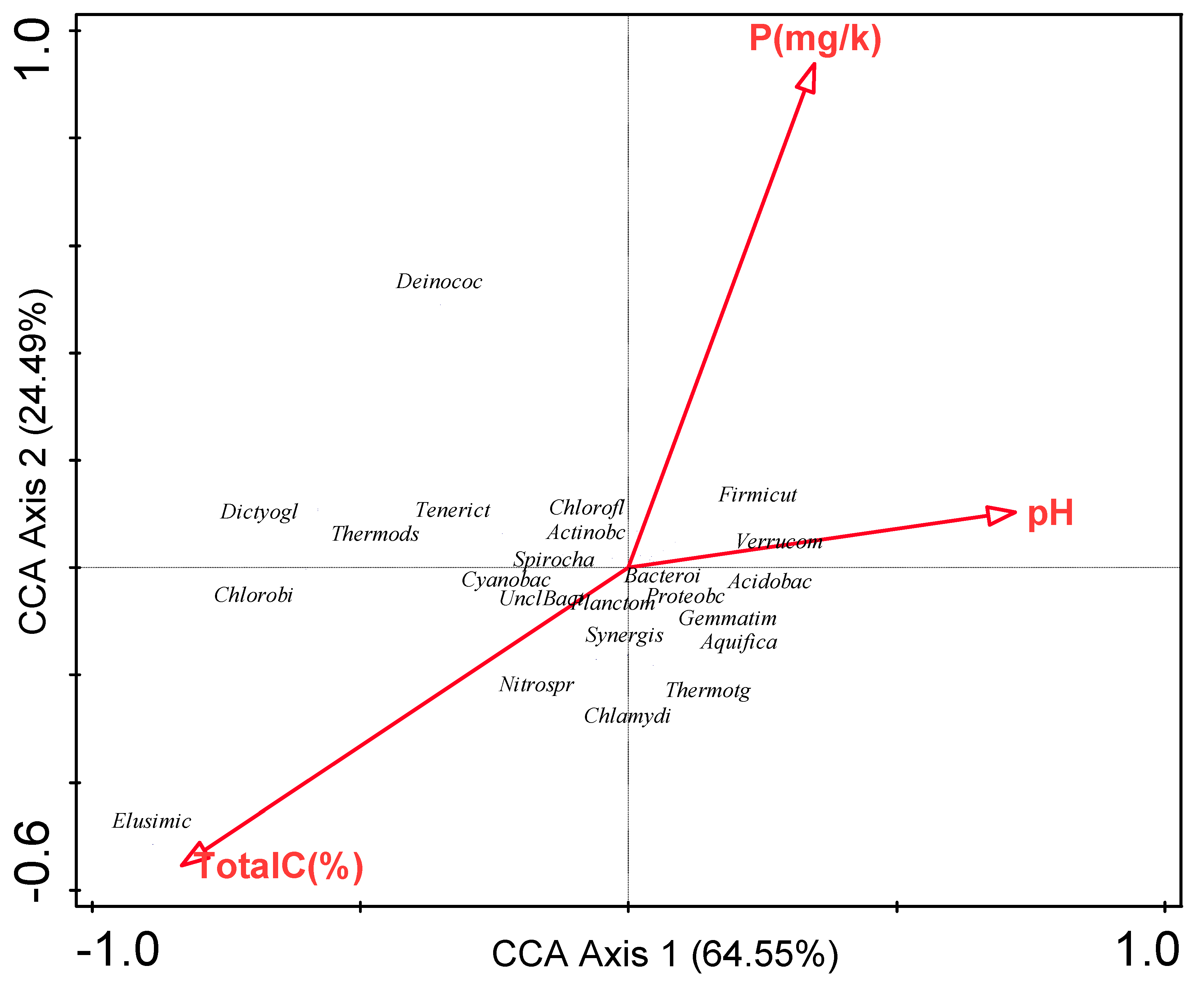
3.6. Influence of Environmental Factors on Bacterial Communities
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Llado, S.; López-Mondéjar, R.; Baldrian, P. Forest soil bacteria: Diversity, involvement in ecosystem processes, and response to global change. Microbiol. Mol. Biol. Rev. 2017, 81, e00063-16. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.A.M.; Lindeskog, M.; Smith, B.; Poulter, B.; Arneth, A.; Haverd, V.; Calle, L. Role of forest regrowth in global carbon sink dynamics. Proc. Natl. Acad. Sci. USA 2019, 116, 4382–4387. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, M.A.; Islam, K.N.; Hafiz, N.; Islam, K. Diversity of trees in a community managed forest: The case of Komolchori VCF, Khagrachari, Bangladesh. Geol. Ecol. Landsc. 2019, 3, 95–103. [Google Scholar] [CrossRef]
- Chodak, M.; Klimek, B.; Niklińska, M. Composition and activity of soil microbial communities in different types of temperate forests. Biol. Fertil. Soils 2016, 52, 1093–1104. [Google Scholar] [CrossRef]
- Baldrian, P.; Kolařík, M.; Štursová, M.; Kopecký, J.; Valášková, V.; Větrovský, T.; Žifčáková, L.; Šnajdr, J.; Rídl, J.; Vlček, Č. Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J. 2012, 6, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Baldrian, P. Forest microbiome: Diversity, complexity and dynamics. FEMS Microbiol. Rev. 2017, 41, 109–130. [Google Scholar] [CrossRef] [PubMed]
- Amoo, A.E.; Babalola, O.O. Ammonia-oxidizing microorganisms: Key players in the promotion of plant growth. J. Soil Sci. Plant Nutr. 2017, 17, 935–947. [Google Scholar] [CrossRef]
- Colombo, F.; Macdonald, C.A.; Jeffries, T.C.; Powell, J.R.; Singh, B.K. Impact of forest management practices on soil bacterial diversity and consequences for soil processes. Soil Biol. Biochem. 2016, 94, 200–210. [Google Scholar] [CrossRef]
- Baldrian, P. Microbial activity and the dynamics of ecosystem processes in forest soils. Curr. Opin. Microbiol. 2017, 37, 128–134. [Google Scholar] [CrossRef]
- Barberán, A.; McGuire, K.L.; Wolf, J.A.; Jones, F.A.; Wright, S.J.; Turner, B.L.; Essene, A.; Hubbell, S.P.; Faircloth, B.C.; Fierer, N. Relating belowground microbial composition to the taxonomic, phylogenetic, and functional trait distributions of trees in a tropical forest. Ecol. Lett. 2015, 18, 1397–1405. [Google Scholar] [CrossRef]
- Kivlin, S.N.; Hawkes, C.V. Temporal and Spatial Variation of Soil Bacteria Richness, Composition, and Function in a Neotropical Rainforest. PLoS ONE 2016, 11, e0159131. [Google Scholar] [CrossRef] [PubMed]
- Szoboszlay, M.; Dohrmann, A.B.; Poeplau, C.; Don, A.; Tebbe, C.C. Impact of land-use change and soil organic carbon quality on microbial diversity in soils across Europe. FEMS Microbiol. Ecol. 2017, 93, fix146. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Zhang, Y.; Qian, W.; Liang, C.; Wang, C.; Tao, S. Land-use changes influence soil bacterial communities in a meadow grassland in Northeast China. Solid Earth 2017, 8, 1119–1129. [Google Scholar] [CrossRef]
- Holland, T.C.; Bowen, P.A.; Bogdanoff, C.P.; Lowery, T.D.; Shaposhnikova, O.; Smith, S.; Hart, M.M. Evaluating the diversity of soil microbial communities in vineyards relative to adjacent native ecosystems. Appl. Soil Ecol. 2016, 100, 91–103. [Google Scholar] [CrossRef]
- Wubie, M.A.; Assen, M.; Nicolau, M.D. Patterns, causes and consequences of land use/cover dynamics in the Gumara watershed of lake Tana basin, Northwestern Ethiopia. Environ. Syst. Res. 2016, 5, 8. [Google Scholar] [CrossRef]
- Liu, J.; Kuang, W.; Zhang, Z.; Xu, X.; Qin, Y.; Ning, J.; Zhou, W.; Zhang, S.; Li, R.; Yan, C. Spatiotemporal characteristics, patterns, and causes of land-use changes in China since the late 1980s. J. Geogr. Sci. 2014, 24, 195–210. [Google Scholar] [CrossRef]
- Amundson, R.; Berhe, A.A.; Hopmans, J.W.; Olson, C.; Sztein, A.E.; Sparks, D.L. Soil and human security in the 21st century. Science 2015, 348, 1261071. [Google Scholar] [CrossRef]
- Foley, J.A.; DeFries, R.; Asner, G.P.; Barford, C.; Bonan, G.; Carpenter, S.R.; Chapin, F.S.; Coe, M.T.; Daily, G.C.; Gibbs, H.K. Global consequences of land use. Science 2005, 309, 570–574. [Google Scholar] [CrossRef]
- Sanderman, J.; Hengl, T.; Fiske, G.J. Soil carbon debt of 12,000 years of human land use. Proc. Natl. Acad. Sci. USA 2017, 114, 9575–9580. [Google Scholar] [CrossRef]
- Ding, G.-C.; Piceno, Y.M.; Heuer, H.; Weinert, N.; Dohrmann, A.B.; Carrillo, A.; Andersen, G.L.; Castellanos, T.; Tebbe, C.C.; Smalla, K. Changes of soil bacterial diversity as a consequence of agricultural land use in a semi-arid ecosystem. PLoS ONE 2013, 8, e59497. [Google Scholar] [CrossRef]
- Delgado-Baquerizo, M.; Oliverio, A.M.; Brewer, T.E.; Benavent-González, A.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K.; Fierer, N. A global atlas of the dominant bacteria found in soil. Science 2018, 359, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.; Lau, S.; Teng, J.; Tse, H.; Yuen, K.-Y. Then and now: Use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin. Microbiol. Infect. 2008, 14, 908–934. [Google Scholar] [CrossRef] [PubMed]
- Enagbonma, B.J.; Aremu, B.R.; Babalola, O.O. Profiling the functional diversity of termite mound soil bacteria as revealed by shotgun sequencing. Genes 2019, 10, 637. [Google Scholar] [CrossRef] [PubMed]
- Santi, C.; Certini, G.; D’Acqui, L.P. Direct determination of organic carbon by dry combustion in soils with carbonates. Commun. Soil Sci. Plant Anal. 2006, 37, 155–162. [Google Scholar] [CrossRef]
- Walkley, A.; Black, I.A. An examination of the Degtjareff method for determining soil organic matter, and a proposed modification of the chromic acid titration method. Soil Sci. 1934, 37, 29–38. [Google Scholar] [CrossRef]
- Deke, A.L.; Adugna, W.T.; Fite, A.T. Soil physic-chemical properties in termite mounds and adjacent control soil in Miyo and Yabello Districts of Borana Zone, Southern Ethiopia. Am. J. Agric. For. 2016, 4, 69–74. [Google Scholar]
- Shi, J.-Y.; Yuan, X.-F.; Lin, H.-R.; Yang, Y.-Q.; Li, Z.-Y. Differences in soil properties and bacterial communities between the rhizosphere and bulk soil and among different production areas of the medicinal plant Fritillaria thunbergii. Int. J. Mol. Sci. 2011, 12, 3770–3785. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef]
- Garcia-Mazcorro, J.F.; Castillo-Carranza, S.A.; Guard, B.; Gomez-Vazquez, J.P.; Dowd, S.E.; Brigthsmith, D.J. Comprehensive molecular characterization of bacterial communities in feces of pet birds using 16S marker sequencing. Microb. Ecol. 2017, 73, 224–235. [Google Scholar] [CrossRef]
- Hammer, Ř.; Harper, D.; Ryan, P. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9–15. [Google Scholar]
- Khomtchouk, B.B.; Hennessy, J.R.; Wahlestedt, C. shinyheatmap: Ultra fast low memory heatmap web interface for big data genomics. PLoS ONE 2017, 12, e0176334. [Google Scholar] [CrossRef] [PubMed]
- Gower, J.C. Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika 1966, 53, 325–338. [Google Scholar] [CrossRef]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral. Ecol. 2001, 26, 32–46. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; Available online: https://www.r-project.org/ (accessed on 26 September 2019).
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A. The metagenomics RAST server–a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faoro, H.; Alves, A.; Souza, E.; Rigo, L.; Cruz, L.; Al-Janabi, S.; Monteiro, R.; Baura, V.; Pedrosa, F. Influence of soil characteristics on the diversity of bacteria in the Southern Brazilian Atlantic Forest. Appl. Environ. Microbiol. 2010, 76, 4744–4749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Gannes, V.; Bekele, I.; Dipchansingh, D.; Wuddivira, M.N.; De Cairies, S.; Boman, M.; Hickey, W.J. Microbial community structure and function of soil following ecosystem conversion from native forests to teak plantation forests. Front. Microbiol. 2016, 7, 1976. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-T.; Jangid, K.; Whitman, W.B.; Coleman, D.C.; Chiu, C.-Y. Change in bacterial community structure in response to disturbance of natural hardwood and secondary coniferous forest soils in central Taiwan. Microb. Ecol. 2011, 61, 429–437. [Google Scholar] [CrossRef]
- Chen, H.; Zhao, X.; Lin, Q.; Li, G.; Kong, W. Using a combination of PLFA and DNA-based sequencing analyses to detect shifts in the soil microbial community composition after a simulated spring precipitation in a semi-arid grassland in China. Sci. Total Environ. 2019, 657, 1237–1245. [Google Scholar] [CrossRef]
- Luo, S.; Wang, S.; Tian, L.; Shi, S.; Xu, S.; Yang, F.; Li, X.; Wang, Z.; Tian, C. Aggregate-related changes in soil microbial communities under different ameliorant applications in saline-sodic soils. Geoderma 2018, 329, 108–117. [Google Scholar] [CrossRef]
- Liu, M.; Sui, X.; Hu, Y.; Feng, F. Microbial community structure and the relationship with soil carbon and nitrogen in an original Korean pine forest of Changbai Mountain, China. BMC Microbiol. 2019, 19, 218. [Google Scholar] [CrossRef] [Green Version]
- Xue, P.-P.; Carrillo, Y.; Pino, V.; Minasny, B.; McBratney, A.B. Soil properties drive microbial community structure in a large scale transect in south eastern Australia. Sci. Rep. 2018, 8, 11725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Wang, C.; Yu, W.; Turak, A.; Chen, D.; Huang, Y.; Ao, J.; Jiang, Y.; Huang, Z. Effects of nitrogen and phosphorus inputs on soil bacterial abundance, diversity, and community composition in Chinese Fir plantations. Front. Microbiol. 2018, 9, 1543. [Google Scholar] [CrossRef] [PubMed]
- Alori, E.T.; Glick, B.R.; Babalola, O.O. Microbial Phosphorus Solubilization and Its Potential for Use in Sustainable Agriculture. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleeson, D.; Mathes, F.; Farrell, M.; Leopold, M. Environmental drivers of soil microbial community structure and function at the Avon River Critical Zone Observatory. Sci. Total Environ. 2016, 571, 1407–1418. [Google Scholar] [CrossRef]
- Tripathi, B.M.; Kim, M.; Lai-Hoe, A.; Shukor, N.A.; Rahim, R.A.; Go, R.; Adams, J.M. pH dominates variation in tropical soil archaeal diversity and community structure. FEMS Microbiol. Ecol. 2013, 86, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, P.; de Miera, L.E.S.; Ansola, G. Influence of environmental variables on the structure and composition of soil bacterial communities in natural and constructed wetlands. Sci. Total Environ. 2015, 506, 380–390. [Google Scholar] [CrossRef]
- Peralta, R.M.; Ahn, C.; Gillevet, P.M. Characterization of soil bacterial community structure and physicochemical properties in created and natural wetlands. Sci. Total Environ. 2013, 443, 725–732. [Google Scholar] [CrossRef]
- Rasche, F.; Knapp, D.; Kaiser, C.; Koranda, M.; Kitzler, B.; Zechmeister-Boltenstern, S.; Richter, A.; Sessitsch, A. Seasonality and resource availability control bacterial and archaeal communities in soils of a temperate beech forest. ISME J. 2011, 5, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Vries, F.T.; Manning, P.; Tallowin, J.R.; Mortimer, S.R.; Pilgrim, E.S.; Harrison, K.A.; Hobbs, P.J.; Quirk, H.; Shipley, B.; Cornelissen, J.H. Abiotic drivers and plant traits explain landscape-scale patterns in soil microbial communities. Ecol. Lett. 2012, 15, 1230–1239. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | TC | TI | WC | WI |
|---|---|---|---|---|
| Organic C (%) | 3.21 ± 0.06 | 4.08 ± 0.07 | 2.97 ± 0.05 | 2.67 ± 0.07 |
| NO3− (mg/kg) | 63.38 ± 0.16 | 65.13 ± 0.01 | 22.01 ± 0.19 | 5.91 ± 0.03 |
| Total C (%) | 3.97 ± 0.05 | 4.58 ± 0.02 | 3.04 ± 0.00 | 2.91 ± 0.03 |
| Total N (%) | 0.01 ± 0.00 | 0.02 ± 0.01 | 0.01 ± 0.00 | 0.001 ± 0.00 |
| pH | 4.28 ± 0.01 | 4.78 ± 0.11 | 5.31 ± 0.03 | 5.10 ± 0.01 |
| P (mg/kg) | 3.16 ± 0.00 | 2.33 ± 0.02 | 3.21 ± 0.08 | 5.03 ± 0.01 |
| Ca (mg/kg) | 15.85 ± 0.05 | 234.50 ± 1.50 | 320.50 ± 1.50 | 164.50 ± 1.50 |
| Mg (mg/kg) | 52.35 ± 0.15 | 95.60 ± 0.10 | 117.00 ± 1.00 | 97.65 ± 0.45 |
| K (mg/kg) | 54.00 ± 0.10 | 70.30 ± 0.10 | 69.95 ± 0.35 | 92.20 ± 0.10 |
| Na (mg/kg) | 11.60 ± 0.30 | 15.35 ± 0.15 | 16.60 ± 0.30 | 15.50 ± 0.20 |
| Sand (%) | 23.50 ± 1.40 | 59.85 ± 3.25 | 55.15 ± 1.95 | 55.10 ± 0.60 |
| Silt (%) | 50.80 ± 1.60 | 35.20 ± 2.10 | 17.45 ± 0.75 | 16.95 ± 0.45 |
| Clay (%) | 19.00 ± 0.10 | 9.00 ± 0.60 | 23.45 ± 1.05 | 25.05 ± 0.75 |
| Bioproject Number | SRR8135323 | SRR8134476 | SRR8136388 | SRR8136221 |
|---|---|---|---|---|
| Sampling site | TC | TI | WC | WI |
| Uploading Information | ||||
| bp count | 41,707,827 | 46,709,377 | 43,701,871 | 66,310,459 |
| Sequences count | 91,160 | 101,138 | 95,076 | 144,319 |
| Mean sequence length (bp) | 458 ± 14 | 458 ± 14 | 460 ± 15 | 459 ± 14 |
| Mean GC content (%) | 57 ± 2 | 57 ± 3 | 56 ± 3 | 56 ± 2 |
| Post QC Information | ||||
| bp count | 5,310,782 | 6,752,690 | 6,060,310 | 7,440,367 |
| Sequences count | 11,570 | 14,666 | 13,134 | 16,140 |
| Mean sequence length (bp) | 459 ± 12 | 460 ± 12 | 461 ± 12 | 461 ± 11 |
| Mean GC content (%) | 57 ± 3 | 57 ± 3 | 56 ± 3 | 56 ± 3 |
| Processed Sequences | ||||
| Predicted protein features | 93 | 85 | 65 | 79 |
| Predicted rRNA features | 14,918 | 22,226 | 16,029 | 26,074 |
| Aligned Sequences | ||||
| Identified protein features | 28 | 27 | 19 | 19 |
| Identified rRNA features | 13,720 | 21,040 | 15,252 | 24,702 |
| Shannon_H | 1.95 | 1.98 | 1.88 | 1.95 |
| Evenness_e^H/S | 1.31 | 1.31 | 1.36 | 1.62 |
| Environmental variable | Explains % | Contribution % | Pseudo-F | P |
|---|---|---|---|---|
| Total C | 52.30 | 52.30 | 2.20 | 0.18 |
| P | 36.80 | 36.80 | 3.40 | 0.22 |
| pH | 11.00 | 11.00 | <0.1 | 1.00 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amoo, A.E.; Babalola, O.O. Impact of Land Use on Bacterial Diversity and Community Structure in Temperate Pine and Indigenous Forest Soils. Diversity 2019, 11, 217. https://doi.org/10.3390/d11110217
Amoo AE, Babalola OO. Impact of Land Use on Bacterial Diversity and Community Structure in Temperate Pine and Indigenous Forest Soils. Diversity. 2019; 11(11):217. https://doi.org/10.3390/d11110217
Chicago/Turabian StyleAmoo, Adenike Eunice, and Olubukola Oluranti Babalola. 2019. "Impact of Land Use on Bacterial Diversity and Community Structure in Temperate Pine and Indigenous Forest Soils" Diversity 11, no. 11: 217. https://doi.org/10.3390/d11110217
APA StyleAmoo, A. E., & Babalola, O. O. (2019). Impact of Land Use on Bacterial Diversity and Community Structure in Temperate Pine and Indigenous Forest Soils. Diversity, 11(11), 217. https://doi.org/10.3390/d11110217