Effectiveness of DNA Barcoding in Speyeria Butterflies at Small Geographic Scales

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
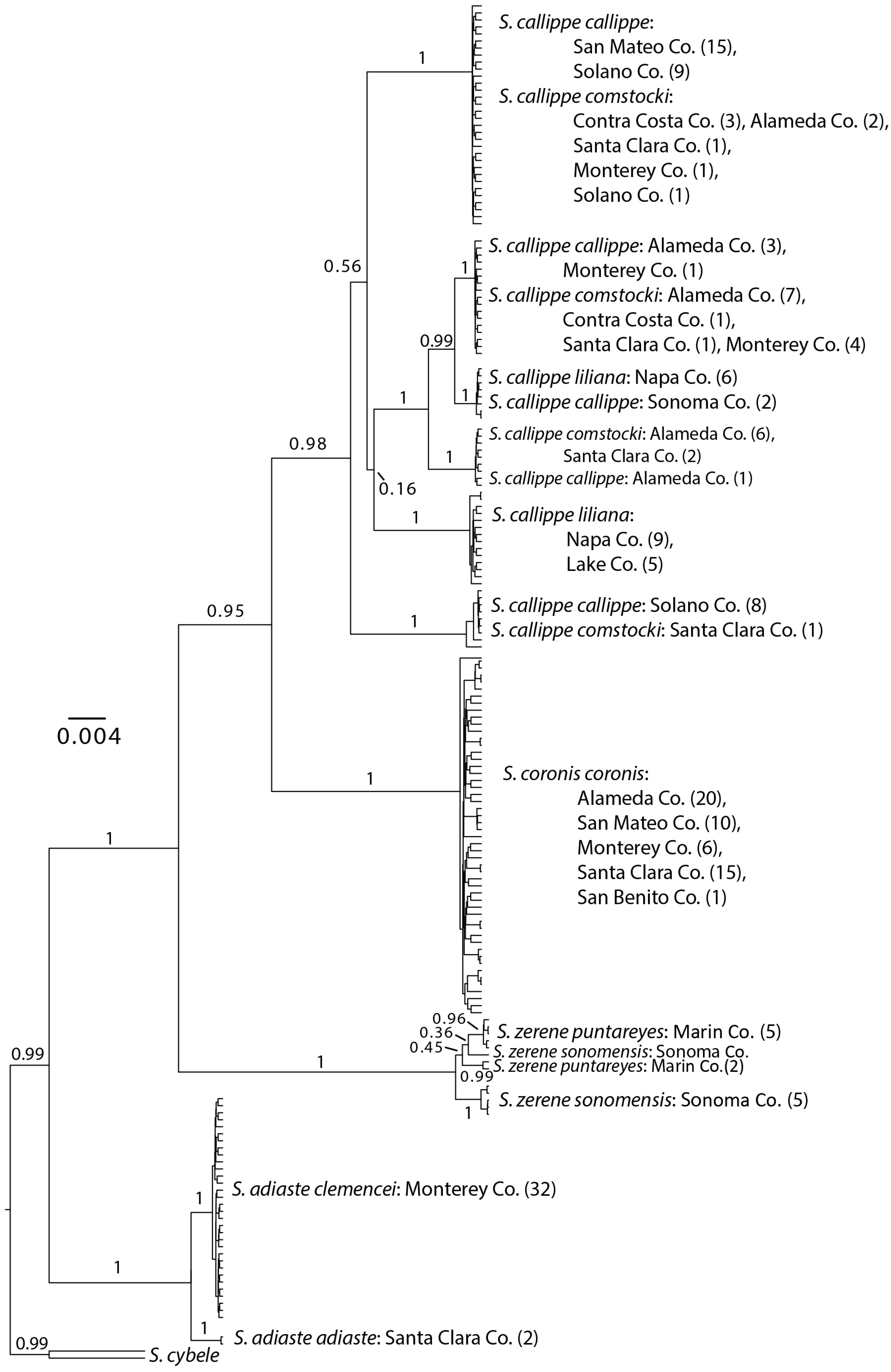
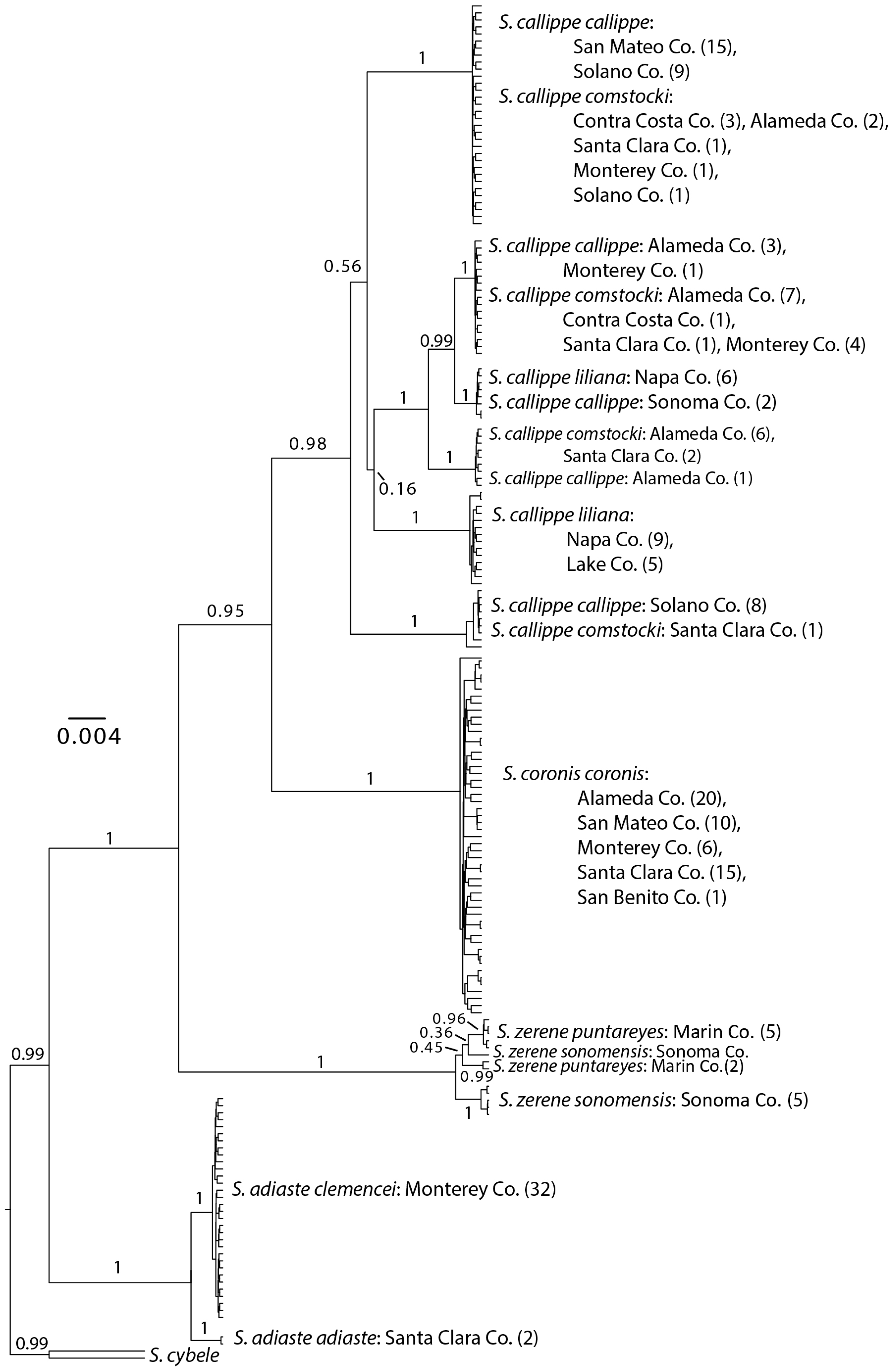
3.1. Neighbor-Joining and Bayesian Results
3.2. Distance Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Emmel, J.F.; Emmel, T.C.; Mattoon, S.O. A new subspecies of Speyeria zerene (Lepidoptera: Nymphalidae) from the San Francisco Bay Area of California. In Systematics of Western North American Butterflies; Emmel, T.C., Ed.; Mariposa Press: Gainesville, FL, USA, 1998; pp. 455–460. [Google Scholar]
- Shapiro, A.M. Montane insular butterfly biogeography: Fauna of Ball Mountain, Siskiyou County, California. Great Basin Nat. 1986, 46, 336–347. [Google Scholar]
- Glassberg, J. Butterflies through Binoculars: The West; Oxford University Press: New York, NY, USA, 2001. [Google Scholar]
- Emmel, T.C.; Austin, G.T. What is Argynnis carolae? The cytotaxonomy and systematic position of a Speyeria relic from Nevada (Lepidoptera: Nymphalidae). In Systematics of Western North American Butterflies; Emmel, T.C., Ed.; Mariposa Press: Gainesville, FL, USA, 1998; pp. 443–450. [Google Scholar]
- McHugh, A.; Bierzychudek, P.; Greever, C.; Marzulla, T.; Van Buskirk, R.; Binford, G. A molecular phylogenetic analysis of Speyeria and its implications for the management of the threatened Speyeria zerene hippolyta. J. Insect Conserv. 2013, 17, 1237–1253. [Google Scholar] [CrossRef]
- Hammond, P.C.; McCorkle, D.V.; Bergman, W. Hybridization studies of genomic compatibility and phenotypic expression in the Greater Fritillary Butterflies (Nymphalidae: Argynnini). J. Lepid. Soc. 2013, 67, 263–273. [Google Scholar] [CrossRef]
- Hammond, P.C. Patterns of geographic variation and evolution in polytypic butterflies. J. Res. Lepid. 1990, 29, 54–76. [Google Scholar]
- De Moya, R.S.; Savage, W.; Tenney, C.; Bao, V.; Wahlberg, N.; Hill, R.I. Interrelationships and diversification of Argynnis Fabricius and Speyeria Scudder butterflies. Syst. Entomol. 2017, 42, 635–649. [Google Scholar] [CrossRef]
- Simonsen, T.J. Fritillary phylogeny, classification, and larval host plants: Reconstructed mainly on the basis of male and female genitalic morphology (Lepidoptera: Nymphalidae: Argynnini). Biol. J. Linn. Soc. 2006, 89, 627–673. [Google Scholar] [CrossRef]
- Simonsen, T.J. Glands, muscles and genitalia. Morphological and phylogenetic implications of histological characters in the male genitalia of Fritillary butterflies (Lepidoptera: Nymphalidae: Argynnini). Zool. Scr. 2006, 35, 231–241. [Google Scholar] [CrossRef]
- Dos Passos, C.F.; Grey, L.P. A genitalic survey of the Argynninae (Lepidoptera, Nymphalidae). Am. Mus Novit. 1945, 1296, 1–29. [Google Scholar]
- Hammond, P.C.; McCorkle, D.V. The decline and extinction of Speyeria populations resulting from human environmental disturbances (Nymphalidae: Argynninae). J. Res. Lepid. 1983, 22, 217–224. [Google Scholar]
- Wells, C.N.; Tonkyn, D.W. Range collapse in the Diana fritillary, Speyeria diana (Nymphalidae). Insect Conserv. Divers. 2014, 7, 365–380. [Google Scholar] [CrossRef]
- Shepherd, S.; Debinski, D. Reintroduction of Regal Fritillary (Speyeria idalia) to a Restored Prairie. Ecol. Restor. 2005, 23, 4. [Google Scholar] [CrossRef]
- Sims, S.R. Speyeria (Lepidoptera: Nymphalidae) Conservation. Insects 2017, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Emmel, J.F.; Emmel, T.C. A previously unrecognized subspecies of Speyeria zerene (Lepidoptera: Nymphalidae) from Coastal Northern California. In Systematics of Western North American Butterflies; Emmel, T.C., Ed.; Mariposa Press: Gainesville, FL, USA, 1998; pp. 451–454. [Google Scholar]
- Dunford, J. The Genus Speyeria and the Speyeria atlantis/Speyeria hesperis Complex: Species and Subspecies Accounts, Systematics, and Biogeography (Lepidoptera: Nymphalidae). Ph.D. Thesis, University of Florida, Gainesville, FL, USA, 2007. [Google Scholar]
- Pelham, J.P. A catalogue of the butterflies of the United States and Canada, with a complete bibliography of the descriptive and systematic literature. J. Res. Lepid. 2008, 40, 1–658. [Google Scholar]
- Hammond, P.C. The colonization of violets and Speyeria butterflies on the ash-pumice fields deposited by Cascadian volcanoes. J. Res. Lepid. 1981, 20, 179–191. [Google Scholar]
- Sperling, F.A.H. Mitochondrial DNA variation and Haldane’s rule in the Papilio glaucus and P. troilus species groups. Heredity 1993, 71, 227–233. [Google Scholar] [CrossRef]
- Dasmahapatra, K.K.; Silva-Vasquez, A.; Chung, J.-W.; Mallet, J. Genetic analysis of a wild-caught hybrid between non-sister Heliconius butterfly species. Biol. Lett. 2007, 3, 660–663. [Google Scholar] [CrossRef]
- Elias, M.; Hill, R.I.; Willmott, K.R.; Dasmahapatra, K.K.; Brower, A.V.Z.; Mallet, J.; Jiggins, C.D. Limited performance of DNA barcoding in a diverse community of tropical butterflies. Proc. R. Soc. B Biol. Sci. 2007, 274, 2881–2889. [Google Scholar] [CrossRef] [Green Version]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- DNAstar. Version 2.1.0.; DNAstar: Maddison, WI, USA, 2011. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Maddison, W.P.; Maddison, D.R. Mesquite: A Modular System for Evolutionary Analysis. Version 2.7.3. 2010. Available online: http://mesquiteproject.org (accessed on 1 January 2017).
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. Public Libr. Sci. Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.O.; Davis, C.S.; Dupuis, J.R.; Muirhead, K.; Sperling, F.A.H. Cross-platform compatibility of de novo-aligned SNPs in a nonmodel butterfly genus. Mol. Ecol. Resour. 2017, 17, e84–e93. [Google Scholar] [CrossRef] [PubMed]
- Zaman, K.; Tenney, C.; Rush, C.E.; Hill, R.I. Population ecology of a California endemic: Speyeria adiaste clemencei. J. Insect Conserv. 2015, 19, 753–763. [Google Scholar] [CrossRef]
- Longcore, T.; Lam, C.S.; Kobernus, P.; Polk, E.; Wilson, J.P. Extracting useful data from imperfect monitoring schemes: Endangered butterflies at San Bruno Mountain, San Mateo County, California (1982–2000) and implications for habitat management. J. Insect Conserv. 2010, 14, 335–346. [Google Scholar] [CrossRef]
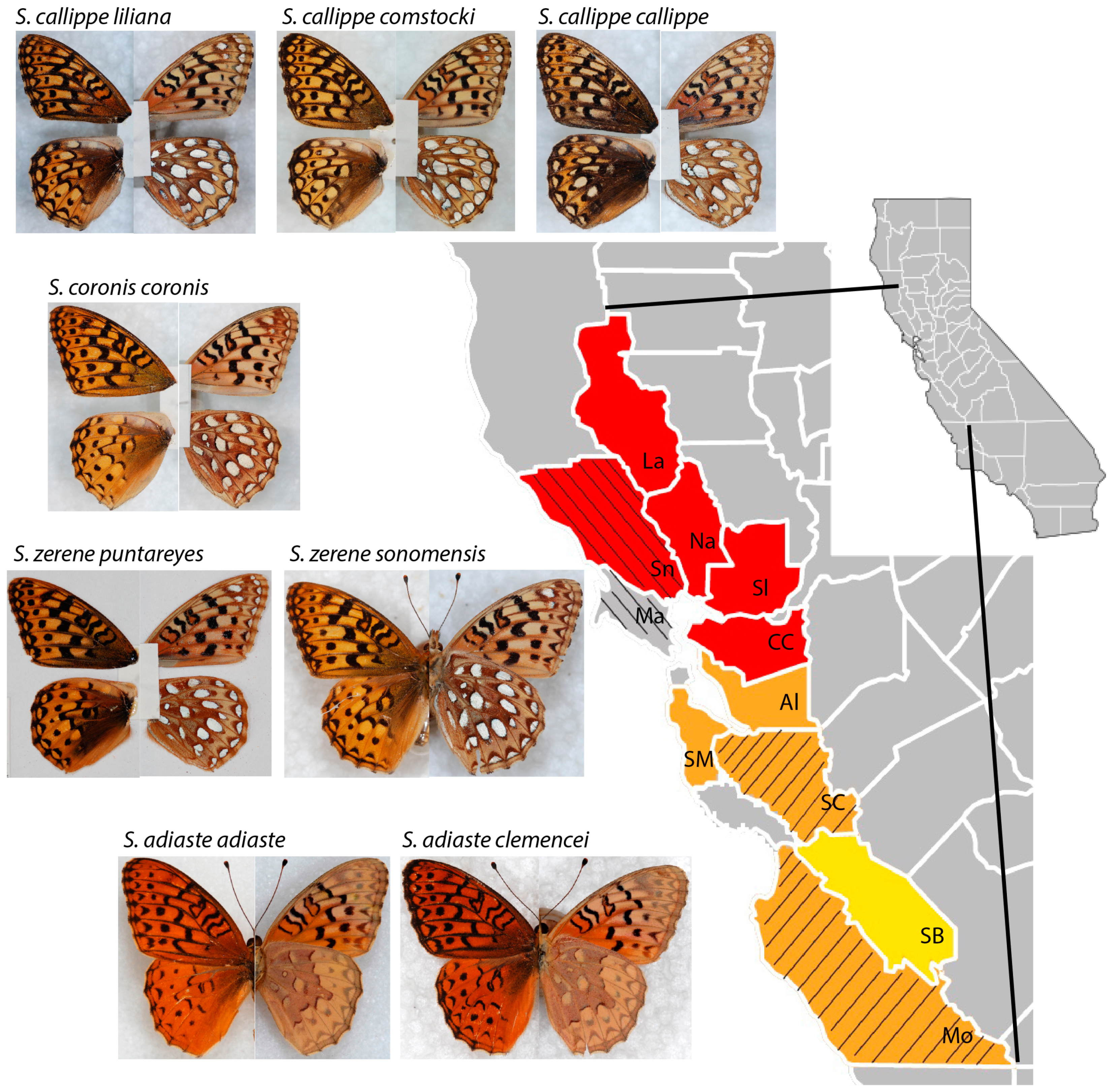

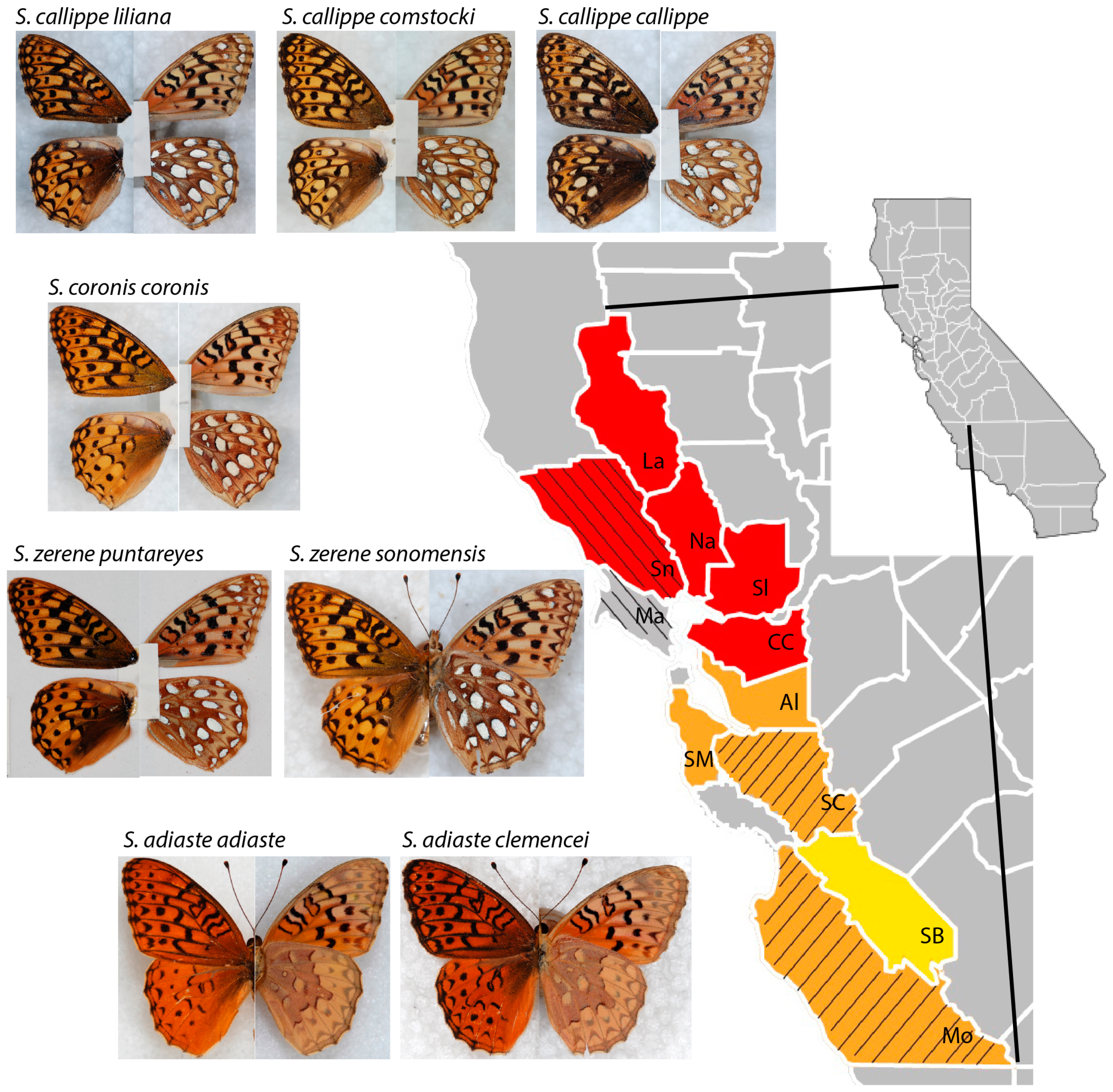{kind=link}
{kind=link}
| S. adiaste | S. callippe | S. coronis | S. zerene | |
|---|---|---|---|---|
| S. adiaste | 0.006 | |||
| S. callippe | 0.031 | 0.028 | ||
| S. coronis | 0.037 | 0.023 | 0.005 | |
| S. zerene | 0.025 | 0.023 | 0.036 | 0.010 |
| S. a. adiaste | S. a. clemencei | S. cal. callippe | S. cal. comstocki | S. cal. liliana | S. cor. coronis | S. z. puntareyes | S. z. sonomensis | |
|---|---|---|---|---|---|---|---|---|
| S. a. adiaste | 0.000 | |||||||
| S. a. clemencei | 0.005 | 0.002 | ||||||
| S. cal. callippe | 0.034 | 0.037 | 0.028 | |||||
| S. cal. comstocki | 0.035 | 0.037 | 0.000 | 0.024 | ||||
| S. cal. liliana | 0.031 | 0.031 | 0.000 | 0.007 | 0.017 | |||
| S. cor. coronis | 0.037 | 0.040 | 0.026 | 0.028 | 0.023 | 0.005 | ||
| S. z. puntareyes | 0.028 | 0.033 | 0.027 | 0.029 | 0.027 | 0.036 | 0.007 | |
| S. z. sonomensis | 0.025 | 0.029 | 0.023 | 0.023 | 0.025 | 0.036 | 0.002 | 0.004 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hill, R.I.; Ganeshan, M.; Wourms, L.; Kronforst, M.R.; Mullen, S.P.; Savage, W.K. Effectiveness of DNA Barcoding in Speyeria Butterflies at Small Geographic Scales. Diversity 2018, 10, 130. https://doi.org/10.3390/d10040130
Hill RI, Ganeshan M, Wourms L, Kronforst MR, Mullen SP, Savage WK. Effectiveness of DNA Barcoding in Speyeria Butterflies at Small Geographic Scales. Diversity. 2018; 10(4):130. https://doi.org/10.3390/d10040130
Chicago/Turabian StyleHill, Ryan I., Maya Ganeshan, Lindsay Wourms, Marcus R. Kronforst, Sean P. Mullen, and Wesley K. Savage. 2018. "Effectiveness of DNA Barcoding in Speyeria Butterflies at Small Geographic Scales" Diversity 10, no. 4: 130. https://doi.org/10.3390/d10040130
APA StyleHill, R. I., Ganeshan, M., Wourms, L., Kronforst, M. R., Mullen, S. P., & Savage, W. K. (2018). Effectiveness of DNA Barcoding in Speyeria Butterflies at Small Geographic Scales. Diversity, 10(4), 130. https://doi.org/10.3390/d10040130