The Molecular Phylogeny of the New Zealand Endemic Genus Hadramphus and the Revival of the Genus Karocolens


Abstract
1. Introduction
2. Methods and Methods
2.1. Specimen Collection
2.2. DNA Extraction, Amplification and Sequencing
2.3. Data Analysis
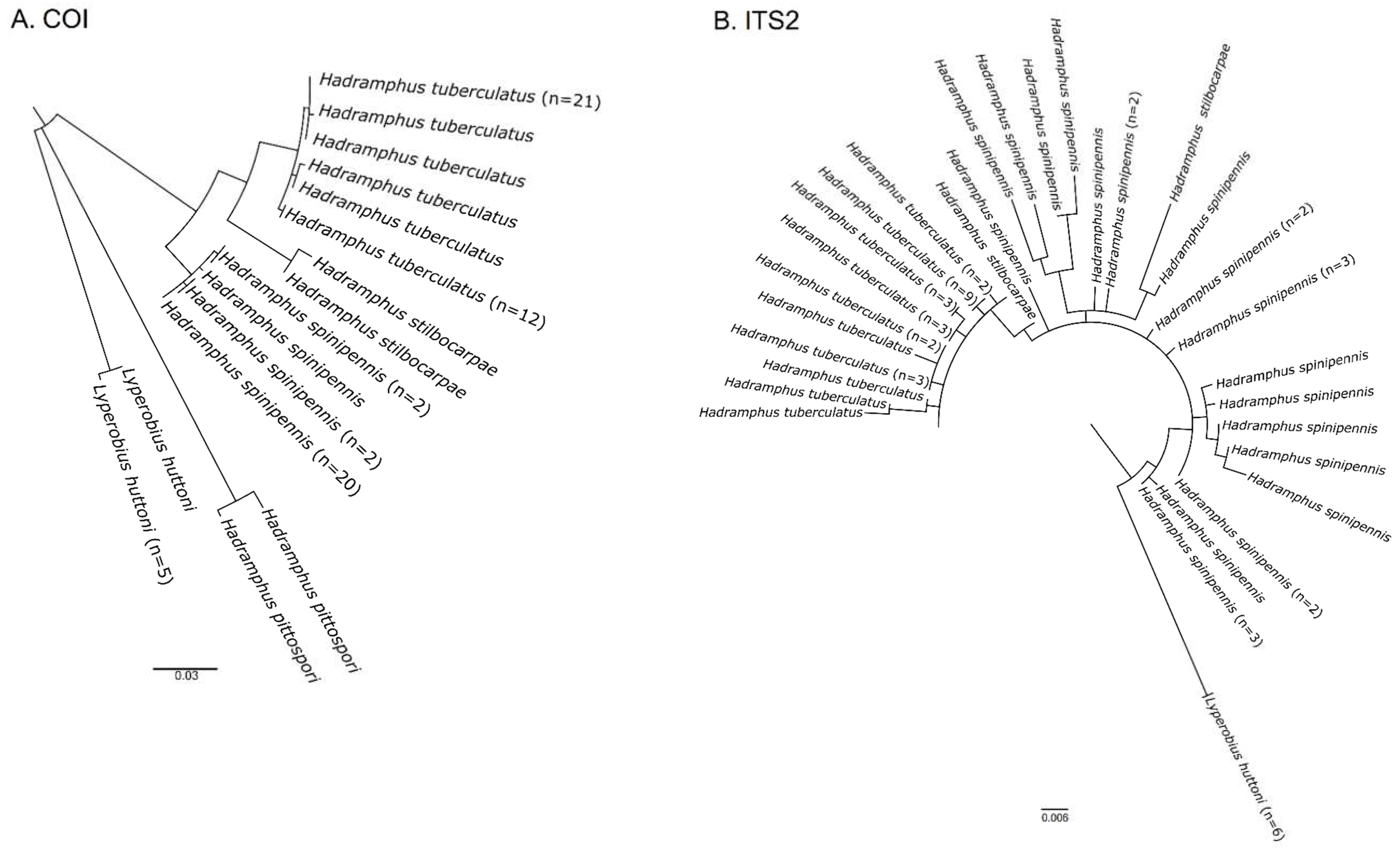
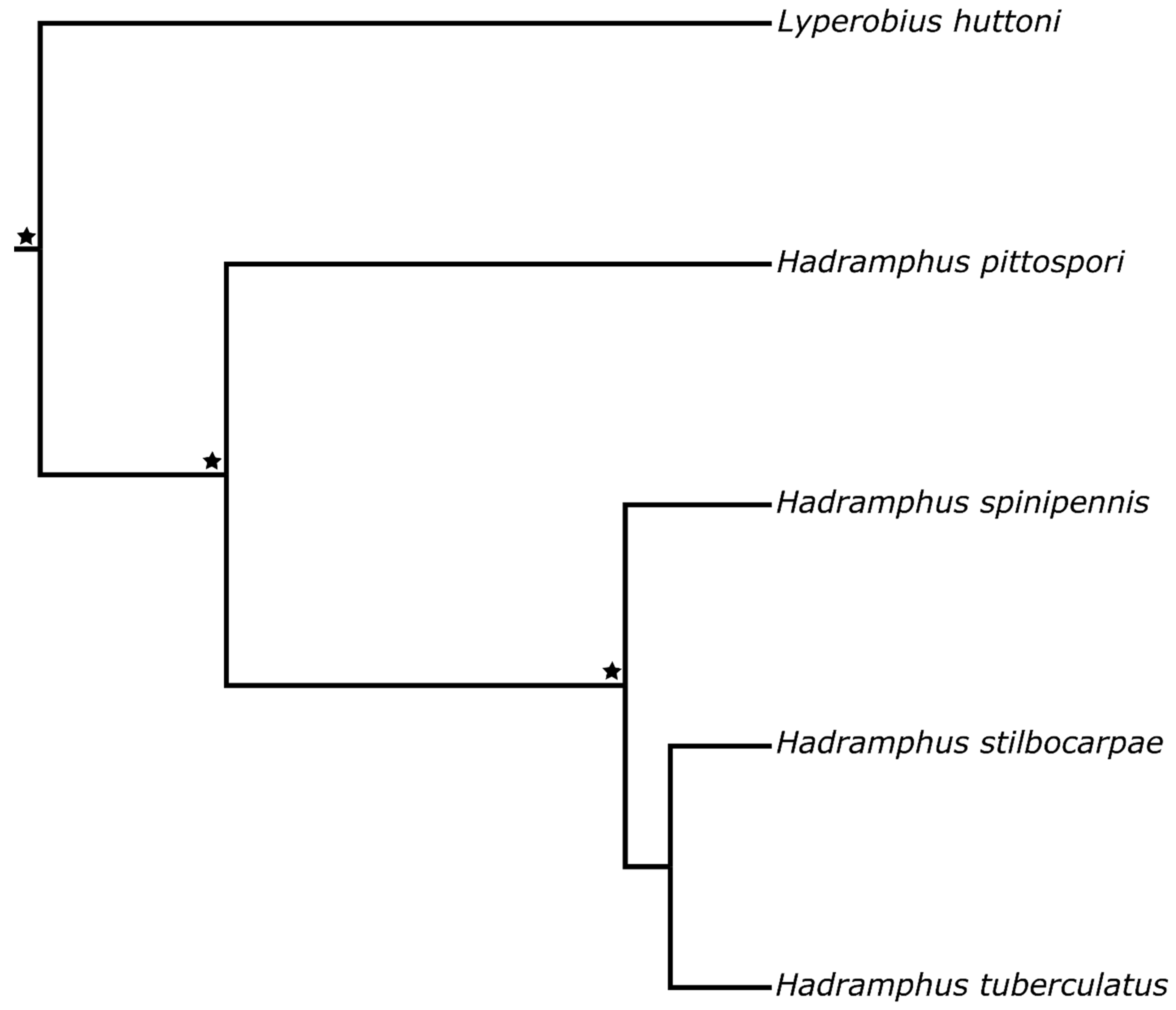
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frankham, R. Challenges and opportunities of genetic approaches to biological conservation. Biol. Conserv. 2010, 143, 1919–1927. [Google Scholar] [CrossRef]
- Yeates, D.K.; Seago, A.; Nelson, L.; Cameron, S.L.; Joseph, L.; Trueman, J.W. Integrative taxonomy, or iterative taxonomy? Syst. Entomol. 2011, 36, 209–217. [Google Scholar] [CrossRef]
- Dayrat, B. Towards integrative taxonomy. Biol. J. Linn. Soc. 2005, 85, 407–415. [Google Scholar] [CrossRef]
- Fujita, M.K.; Leaché, A.D.; Burbrink, F.T.; McGuire, J.A.; Moritz, C. Coalescent-based species delimitation in an integrative taxonomy. Trends Ecol. Evol. 2012, 27, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Scotland, R.W.; Olmstead, R.G.; Bennett, J.R. Phylogeny reconstruction: The role of morphology. Syst. Biol. 2003, 52, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Vink, C.J.; Sirvid, P.J.; Malumbres-Olarte, J.; Griffiths, J.W.; Paquin, P.; Paterson, A.M. Species status and conservation issues of New Zealand’s endemic latrodectus spider species (Araneae: Theridiidae). Invertebr. Syst. 2008, 22, 589–604. [Google Scholar] [CrossRef]
- Edwards, S.V. Is a new and general theory of molecular systematics emerging? Evolution 2009, 63, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, J.; Knowles, L.L. Multispecies coalescent delimits structure, not species. Proc. Natl. Acad. Sci. USA 2017, 114, 1607–1612. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.P.; Fernández, R.; Esposito, L.A.; González-Santillán, E.; Monod, L. Phylogenomic resolution of scorpions reveals multilevel discordance with morphological phylogenetic signal. Proc. R. Soc. Lond. B Biol. Sci. 2015, 282, 20142953. [Google Scholar] [CrossRef] [PubMed]
- Vuataz, L.; Sartori, M.; Gattolliat, J.-L.; Monaghan, M.T. Endemism and diversification in freshwater insects of madagascar revealed by coalescent and phylogenetic analysis of museum and field collections. Mol. Phylogen. Evol. 2013, 66, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Rutschmann, S.; Gattolliat, J.L.; Hughes, S.J.; Báez, M.; Sartori, M.; Monaghan, M.T. Evolution and island endemism of morphologically cryptic baetis and cloeon species (ephemeroptera, baetidae) on the canary islands and madeira. Fresh. Biol. 2014, 59, 2516–2527. [Google Scholar] [CrossRef]
- Vitecek, S.; Kučinić, M.; Previšić, A.; Živić, I.; Stojanović, K.; Keresztes, L.; Bálint, M.; Hoppeler, F.; Waringer, J.; Graf, W. Integrative taxonomy by molecular species delimitation: Multi-locus data corroborate a new species of Balkan Drusinae micro-endemics. BMC Evol. Biol. 2017, 17, 129. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, M.; Knox, G.; Burrows, C.; Marsden, I. The Natural History of Canterbury, 3rd ed.; Canterbury University Press: Christchurch NZ, New Zealand, 2008; p. 923. [Google Scholar]
- Cooper, R.A.; Millener, P.R. The New Zealand biota-historical background and new research. Trends Ecol. Evol. 1993, 8, 429–433. [Google Scholar] [CrossRef]
- Caldecott, J.O.; Jenkins, M.D.; Johnson, T.H.; Groombridge, B. Priorities for conserving global species richness and endemism. Biodivers. Conserv. 1996, 5, 699–727. [Google Scholar] [CrossRef]
- Gillespie, R.G.; Roderick, G.K. Arthropods on islands: Colonization, speciation, and conservation. Annu. Rev. Entomol. 2002, 47, 595–632. [Google Scholar] [CrossRef] [PubMed]
- Myers, N.; Mittermeier, R.A.; Mittermeier, C.G.; Da Fonseca, G.A.; Kent, J. Biodiversity hotspots for conservation priorities. Nature 2000, 403, 853. [Google Scholar] [CrossRef] [PubMed]
- Craw, R.C. Fauna of New Zealand: Molytini Number 39; Manaaki Whenua Press: Lincoln, Canterbury, New Zealand, 1999; p. 68. [Google Scholar]
- Young, L.M.; Marris, J.W.M.; Pawson, S. Back from extinction: Rediscovery of the canterbury knobbled weevil Hadramphus tuberculatus (pascoe 1877) (Coleoptera: Curculionidae), with a review of its historical distribution. N. Z. J. Zool. 2008, 35, 323–330. [Google Scholar] [CrossRef]
- Kuschel, G.; Worthy, T.H. Past distribution of large weevils (Coleoptera: Curculionidae) in the South Island, New Zealand, based on holocene fossil remains. N. Z. Entomol. 1996, 19, 15–22. [Google Scholar] [CrossRef]
- Fountain, E.D.; Wiseman, B.H.; Cruickshank, R.H.; Paterson, A.M. The ecology and conservation of Hadramphus tuberculatus (pascoe 1877) (Coleoptera: Curculionidae: Molytinae). J. Insect Conserv. 2013, 17, 737–745. [Google Scholar] [CrossRef]
- Fountain, E.D.; Pugh, A.R.; Wiseman, B.H.; Smith, V.R.; Cruickshank, R.H.; Paterson, A.M. Captive rearing of the endangered weevil Hadramphus tuberculatus (pascoe, 1877) (Coleoptera: Curculionidae: Molytinae) for ex-situ conservation. N. Z. Entomol. 2016, 39, 23–32. [Google Scholar] [CrossRef]
- Schöps, K.; Wratten, S.D.; Emberson, R.M. Life cycle, behaviour and conservation of the large endemic weevil, Hadramphus spinipennis on the Chatham Islands, New Zealand. N. Z. J. Zool. 1999, 26, 55–66. [Google Scholar] [CrossRef][Green Version]
- Fountain, E.D.; Malumbres-Olarte, J.; Cruickshank, R.H.; Paterson, A.M. The effects of island forest restoration on open habitat specialists: The endangered weevil hadramphus spinipennis broun and its host-plant Aciphylla dieffenbachii kirk. PeerJ 2015, 3, e749. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.W.; Meads, M.; Notman, P.R. A Report on the Restoration of Knobbled Weevils (Hadramphus stilbocarpae) and Flax Weevils (Anagotus fairburni) to Breaksea Island, Breaksea Sound, Fiordland; DSIR Land Resources: Lower Hutt, New Zealand, 1992; p. 30. [Google Scholar]
- Kuschel, G. The subfamily Molytinae (Coleoptera: Curculionidae): General notes and descriptions of new taxa from New Zealand and Chile. N. Z. Entomol. 1987, 9, 11–29. [Google Scholar] [CrossRef]
- Goldberg, J.; Trewick, S.A. Exploring phylogeographic congruence in a continental island system. Insects 2011, 2, 369–399. [Google Scholar] [CrossRef] [PubMed]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Hajibabaei, M.; Janzen, D.H.; Burns, J.M.; Hallwachs, W.; Hebert, P.D.N. DNA barcodes distinguish species of tropical lepidoptera. Proc. Natl. Acad. Sci. USA 2006, 103, 968–971. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T. Ncbi blast: A better web interface. Nucleic Acids Res. 2008, 36, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Löytynoja, A.; Goldman, N. Webprank: A phylogeny-aware multiple sequence aligner with interactive alignment browser. BMC Bioinform. 2010, 11, 579. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. Dnasp 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. Jmodeltest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Zharkikh, A. Estimation of evolutionary distances between nucleotide sequences. J. Mol. Evol. 1994, 39, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, H.A.; Drummond, A.J. StarBEAST2 brings faster species tree inference and accurate estimates of substitution rates. Mol. Biol. Evol. 2017, 34, 2101–2114. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.R.; Drummond, A.J. Bmodeltest: Bayesian phylogenetic site model averaging and model comparison. BMC Evol. Biol. 2017, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Suchard, M.A.; Xie, D.; Drummond, A. Tracer v1.6. 2014. Available online: http://beast.bio.ed.ac.uk/Tracer.
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. Figtree v1. 4. Molecular Evolution, Phylogenetics and Epidemiology; University of Edinburgh: Edinburgh, UK, 2012. [Google Scholar]
- Kuschel, G. Entomology of the Aucklands and other Islands South of New Zealand: Coleoptera: Cuculionidae. Pac. Insects Monogr. 1971, 27, 225–259. [Google Scholar]
- Lewis, P.O. A likelihood approach to estimating phylogeny from discrete morphological character data. Syst. Biol. 2001, 50, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, A.; Anastasiou, I.; Vogler, A.P. Revisiting the insect mitochondrial molecular clock: The mid-aegean trench calibration. Mol. Biol. Evol. 2010, 27, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.G. Observations on rearing Karocolens pittospori (Coleoptera: Curculionidae: Molytinae). N. Z. Entomol. 1987, 9, 34–37. [Google Scholar] [CrossRef]
- Gilbert, M.T.P.; Bandelt, H.-J.; Hofreiter, M.; Barnes, I. Assessing ancient DNA studies. Trends Ecol. Evol. 2005, 20, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, A.S.; Stepien, C.C.; Sijapati, M.; Roque Albelo, L. Comparative genetic structure and demographic history in endemic galápagos weevils. J. Hered. 2012, 103, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Hickerson, M.J.; Meyer, C.P.; Moritz, C. DNA barcoding will often fail to discover new animal species over broad parameter space. Syst. Biol. 2006, 55, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Hendrich, L.; Pons, J.; Ribera, I.; Balke, M. Mitochondrial cox1 sequence data reliably uncover patterns of insect diversity but suffer from high lineage-idiosyncratic error rates. PLoS ONE 2011, 5, e14448. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.Y.; Lo, N. The insect molecular clock. Aust. J. Entomol. 2013, 52, 101–105. [Google Scholar] [CrossRef]
- Gunter, N.L.; Oberprieler, R.G.; Cameron, S.L. Molecular phylogenetics of Australian weevils (Coleoptera: Curculionoidea): Exploring relationships in a hyperdiverse lineage through comparison of independent analyses. Aust. Entomol. 2016, 55, 217–233. [Google Scholar] [CrossRef]
- Queiroz, K.D. Including the characters of interest during tree reconstruction and the problems of circularity and bias in studies of character evolution. Am. Nat. 1996, 148, 700–708. [Google Scholar] [CrossRef]
- Emerson, B.C.; Wallis, G.P. Phylogenetic relationships of the prodontria (Coleoptera; Scarabaeidae; subfamily Melolonthinae), derived from sequence variation in the mitochondrial cytochrome oxidase II gene. Mol. Phylogen. Evol. 1995, 4, 433–447. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Species | N | # Haplotypes | # Polymorphic Sites | Π (STD) |
|---|---|---|---|---|
| huttoni | 10 | 2 | 1 | 0.001 (0.0002) |
| pittospori | 2 | 2 | 5 | 0.012 (0.0059) |
| spinipennis | 28 | 4 | 3 | 0.001 (0.0003) |
| tuberculatus | 71 | 7 | 6 | 0.002 (0.0003) |
| stilbocarpae | 2 | 2 | 3 | 0.007 (0.0035) |
| Species | huttoni | pittospori | spinipennis | tuberculatus | stilbocarpae |
|---|---|---|---|---|---|
| huttoni | 0.001 | 0.029 | 0.024 | 0.024 | 0.025 |
| pittospori | 0.289 | 0.010 | 0.027 | 0.028 | 0.028 |
| spinipennis | 0.210 | 0.270 | 0.007 | 0.011 | 0.013 |
| tuberculatus | 0.210 | 0.270 | 0.052 | 0.002 | 0.012 |
| stilbocarpae | 0.210 | 0.287 | 0.071 | 0.061 | 0.001 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fountain, E.D.; Cruickshank, R.H.; Paterson, A.M. The Molecular Phylogeny of the New Zealand Endemic Genus Hadramphus and the Revival of the Genus Karocolens. Diversity 2018, 10, 88. https://doi.org/10.3390/d10030088
Fountain ED, Cruickshank RH, Paterson AM. The Molecular Phylogeny of the New Zealand Endemic Genus Hadramphus and the Revival of the Genus Karocolens. Diversity. 2018; 10(3):88. https://doi.org/10.3390/d10030088
Chicago/Turabian StyleFountain, Emily D., Robert H. Cruickshank, and Adrian M. Paterson. 2018. "The Molecular Phylogeny of the New Zealand Endemic Genus Hadramphus and the Revival of the Genus Karocolens" Diversity 10, no. 3: 88. https://doi.org/10.3390/d10030088
APA StyleFountain, E. D., Cruickshank, R. H., & Paterson, A. M. (2018). The Molecular Phylogeny of the New Zealand Endemic Genus Hadramphus and the Revival of the Genus Karocolens. Diversity, 10(3), 88. https://doi.org/10.3390/d10030088