The Evolution and Population Diversity of Bison in Pleistocene and Holocene Eurasia: Sex Matters


 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Morphometric Analysis of Bison Metapodials
Source of Osteometric Measurements of Bison Samples
2.2. Analyses of Osteometric Measurements
2.3. Phylogenetic Analyses of Published Bison Mitogenomes
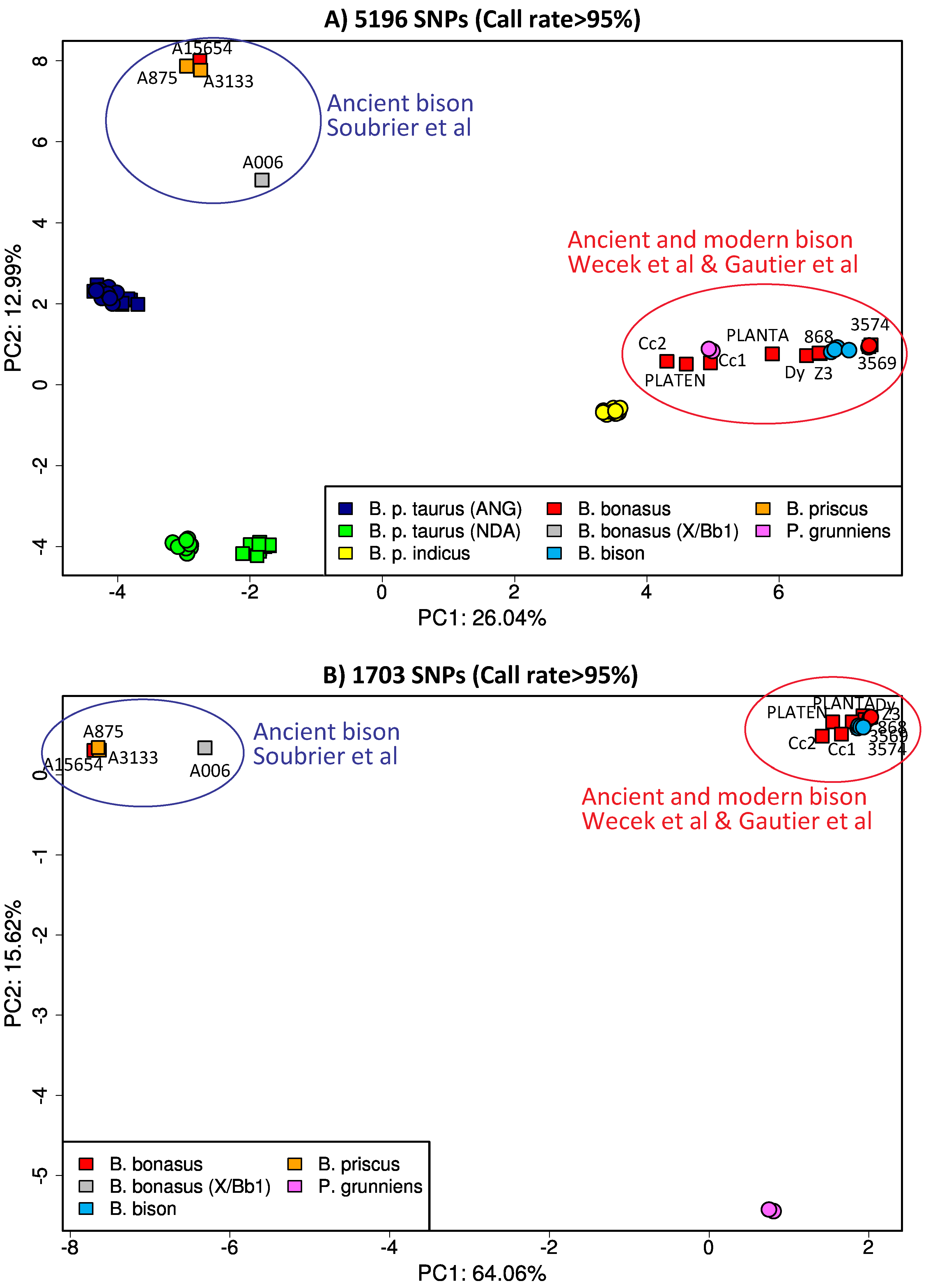
2.4. Meta-Analysis of Publicly Available Genotyping Data Using PCA
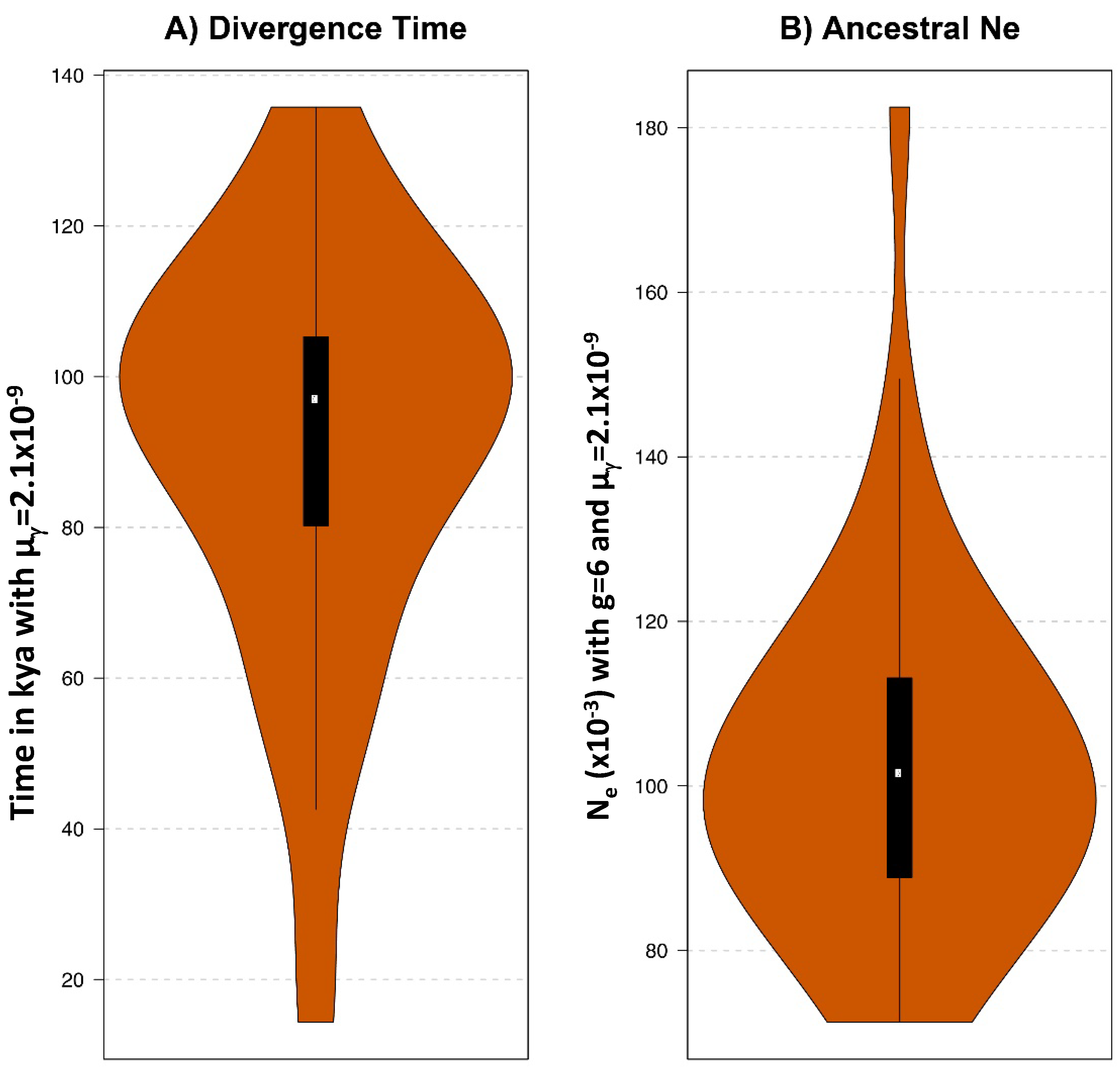
2.5. Estimation of the Divergence between European and American Bison under the CoalHMM Framework
3. Results and Discussion
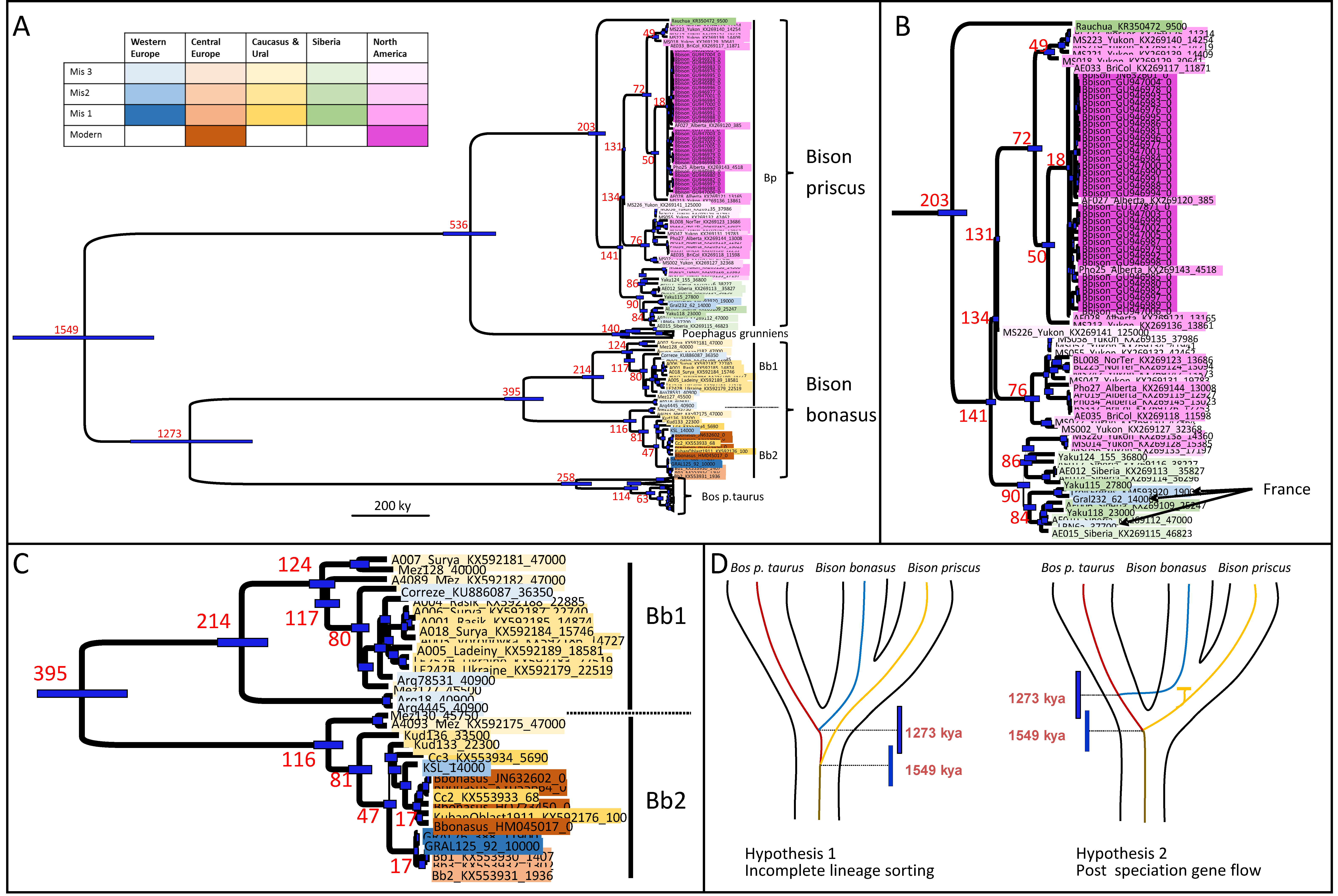
3.1. Phylogenetic Analyses of Ancient Bison Mitogenomes
3.2. Bison Priscus in Eurasia and Its Expansion in America
3.3. Bison Bonasus Origin and Evolution
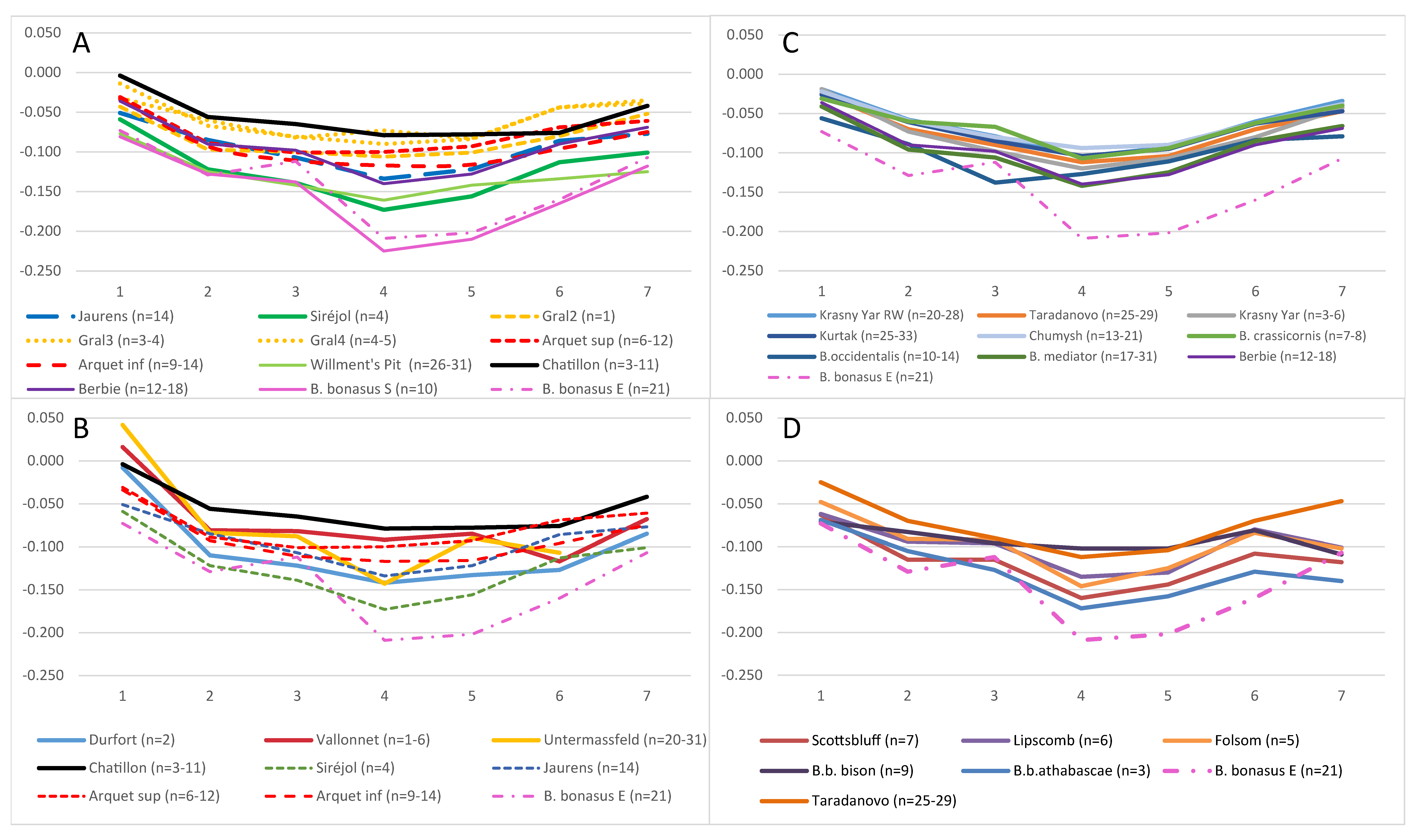
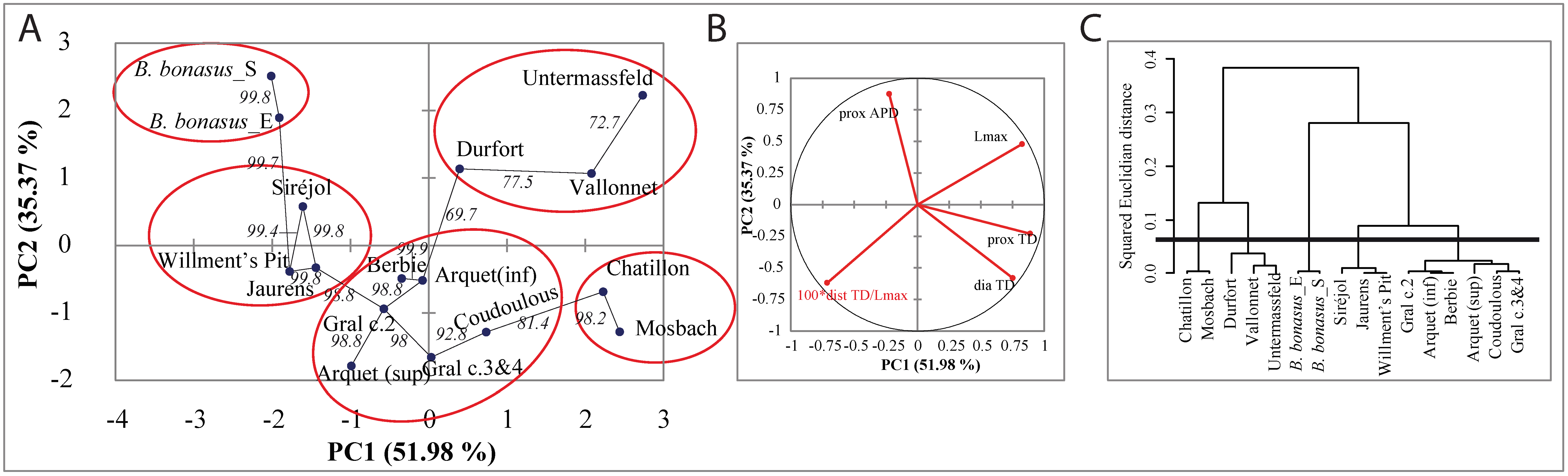
3.4. Morphometric Analysis of Bison Metapodials
3.5. Comparative Genomic Analyses of Ancient and Modern Bison
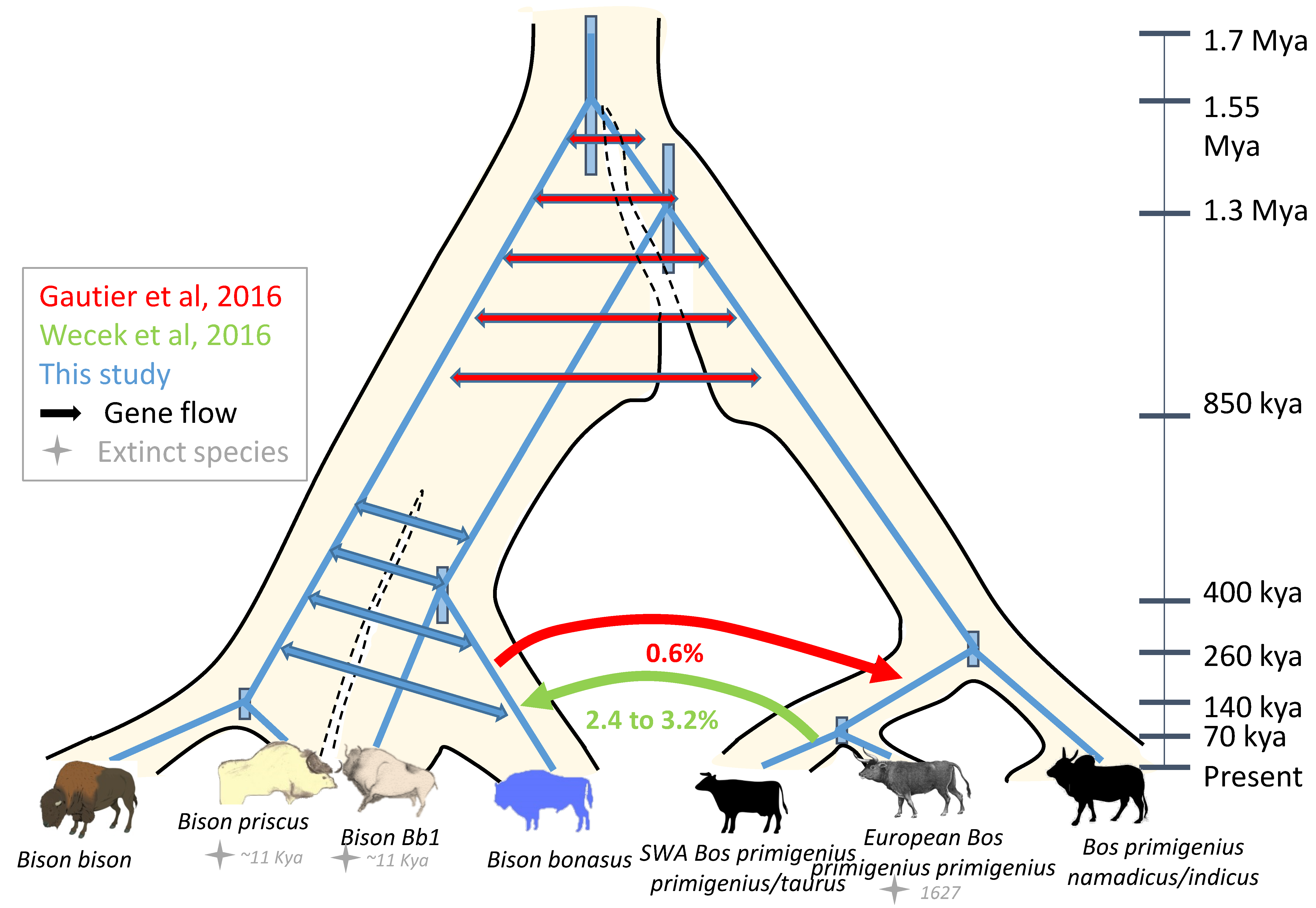
3.6. Integration of the Paleontological, Morphometrics, Genomic and Mitogenomic Data
3.6.1. Morphometric Data and Mitogenome Lineages
3.6.2. Mitogenome Lineages and Nuclear Genomes
3.6.3. Sex-Specific Differences
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Froese, D.; Stiller, M.; Heintzman, P.D.; Reyes, A.V.; Zazula, G.D.; Soares, A.E.; Meyer, M.; Hall, E.; Jensen, B.J.; Arnold, L.J.; et al. Fossil and genomic evidence constrains the timing of bison arrival in North America. Proc. Natl. Acad. Sci. USA 2017, 114, 3457–3462. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, B.; Drummond, A.J.; Rambaut, A.; Wilson, M.C.; Matheus, P.E.; Sher, A.V.; Pybus, O.G.; Gilbert, M.T.; Barnes, I.; Binladen, J.; et al. Rise and fall of the Beringian steppe bison. Science 2004, 306, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Flerow, C.C. Systematics and evolution. In European Bison—Morphology, Systematics, Evolution, Ecology (in Russian); Sokolov, V.E., Ed.; Nauka (USSR Ac. of Sc.): Moscou, Russia, 1979; pp. 9–127. [Google Scholar]
- Sher, A.V. An early quaternary bison population from untermassfeld: Bison menneri sp. nov. In Das Pleistozän von Untermassfeld bei Meiningen (Thüringen); Kahlke, R.D., Ed.; Römisch-Germanisches Zentralmuseum, Leibniz-Forschungsinstitut für Archäologie, Arbeitsbereich Wissenschaftlicher Verlag: Mainz, Germany, 1997; pp. 101–180. [Google Scholar]
- Drees, M. Sexual dimorphism in Pleistocene Bison priscus (Mammalia, Bovidae) with a discussion on the position of Bison schoetensacki. Senckenberg. Lethaea 2005, 85, 153–157. [Google Scholar] [CrossRef]
- Sala, B. Bison schoetensacki Freud. From Isernia la Pineta (early Mid-Pleistocene, Italy) and revision of the european species of bison. Palaeontogr. Ital. 1986, 74, 113–170. [Google Scholar]
- Gautier, M.; Moazami-Goudarzi, K.; Leveziel, H.; Parinello, H.; Groh, C.; Riall, S.; Kowalczyk, R.; Flori, L. Deciphering the wisent demographic and adaptive histories from individual whole-genome sequences. Mol. Biol. Evol. 2016, 33, 2801–2814. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, L.; Lenstra, J.A.; Jian, J.; Yang, Y.; Hu, Q.; Lai, D.; Qiu, Q.; Ma, T.; Du, Z.; et al. The genome sequence of the wisent (Bison bonasus). GigaScience 2017, 6, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Wecek, K.; Hartmann, S.; Paijmans, J.L.A.; Taron, U.; Xenikoudakis, G.; Cahill, J.A.; Heintzman, P.D.; Shapiro, B.; Baryshnikov, G.; Bunevich, A.N.; et al. Complex admixture preceded and followed the extinction of wisent in the wild. Mol. Biol. Evol. 2016, 34, 598–612. [Google Scholar]
- Massilani, D.; Guimaraes, S.; Brugal, J.P.; Bennett, E.A.; Tokarska, M.; Arbogast, R.M.; Baryshnikov, G.; Boeskorov, G.; Castel, J.C.; Davydov, S.; et al. Past climate changes, population dynamics and the origin of Bison in Europe. BMC Biol. 2016, 14, 93. [Google Scholar] [CrossRef] [PubMed]
- Soubrier, J.; Gower, G.; Chen, K.; Richards, S.M.; Llamas, B.; Mitchell, K.J.; Ho, S.Y.; Kosintsev, P.; Lee, M.S.; Baryshnikov, G.; et al. Early cave art and ancient DNA record the origin of European bison. Nat. Commun. 2016, 7, 13158. [Google Scholar] [CrossRef] [PubMed]
- Palacio, P.; Berthonaud, V.; Guerin, C.; Lambourdiere, J.; Maksud, F.; Philippe, M.; Plaire, D.; Stafford, T.; Marsolier-Kergoat, M.C.; Elalouf, J.M. Genome data on the extinct Bison schoetensacki establish it as a sister species of the extant European bison (Bison bonasus). BMC Evol. Biol. 2017, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Lenstra, J.A.; Liu, J. The year of the wisent. BMC Biol. 2016, 14, 100. [Google Scholar] [CrossRef] [PubMed]
- Hassanin, A.; An, J.; Ropiquet, A.; Nguyen, T.T.; Couloux, A. Combining multiple autosomal introns for studying shallow phylogeny and taxonomy of Laurasiatherian mammals: Application to the tribe Bovini (Cetartiodactyla, Bovidae). Mol. Phylogenet. Evol. 2013, 66, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Verkaar, E.L.; Nijman, I.J.; Beeke, M.; Hanekamp, E.; Lenstra, J.A. Maternal and paternal lineages in cross-breeding bovine species. Has wisent a hybrid origin? Mol. Biol. Evol. 2004, 21, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Bedord, J.N. A technique for sex determination of mature bison metapodials. Plains Anthropol. 1978, 23, 40–43. [Google Scholar] [CrossRef]
- Brugal, J.-P. Le Bos primigenius Boj, 1827 du Pléistocène moyen des grottes de Lunel-Viel (Hérault). Bull. Mus. Anthropol. Préhist. Monaco Monaco 1985, 28, 7–62. [Google Scholar]
- Lewis, P.J.; Buchanan, B.; Johnson, E. Sexing bison metapodials using principal componentn analysis. Plains Anthropol. 2005, 50, 1–14. [Google Scholar] [CrossRef]
- Kobrynczuk, F.; Krasinska, M.; Szara, T. Sexual dimorphism in skulls of the lowland European bison, Bison bonasus bonasus. Ann. Zool. Fenn. 2008, 45, 335–340. [Google Scholar] [CrossRef]
- Brugal, J.P. Le bison (Bovinae, Artiodactyla) du gisement pléistocène moyen ancien de durfort (Gard, France). Bull. Mus. Natl. Hist. Nat. Paris 1995, 16, 349–381. [Google Scholar]
- Schertz, E. Der geschlechtsunterschied an metapodien von bison. Senckenberg. Lethaea 1936, 18, 357-38. [Google Scholar]
- Mourer-Chauvire, C. Etude de nouveaux restes de vertébrés provenant de la carrière Fournier à Chatillon-Saint-Jean (Drôme). III: Artiodactyles, chevaux et oiseaux. Bull. Assoc. Fr. Etud. Quat. 1972, 4, 271–302. [Google Scholar]
- Guérin, C.; Valli, A.M.F. Le gisement pléistocène supérieur de la grotte de Jaurens à Nespouls, Corrèze: Les Bovidae (Mammalia, Artiodactyla). Cah. Sci. Mus. Hist. Nat. Lyon 2000, 1, 7–39. [Google Scholar]
- Vasiliev, S.K. Late Pleistocene bison (Bison p. priscus bojanus, 1827) from the southeastern part of Western Siberia. Archaeol. Ethnol. Anthropol. Eurasia 2008, 34, 34–56. [Google Scholar] [CrossRef]
- Empel, W.; Roskosz, T. Das skelett der gliedmaβen des wisents, Bison bonasus linnaeus 1758. Acta Theriol. 1963, 7, 259–300. [Google Scholar] [CrossRef]
- Meadow, R.H. Early animal domestication in South Asia a first report of the faunal remains from mehrgarh pakistan. In South Asian Archaeology 1979; Härtel, H., Ed.; Dietrich Reimer: Berlin, Germany, 1981; pp. 143–179. [Google Scholar]
- Meadow, R.H. The use of size index scaling techniques for research on archaeozoological collections from the Middle East. In Historici Animalium ex Ossibus. Festschrift Angela Von Den Driesch Zum 65. Geburtstag; Becker, C., Manhart, H., Peters, J., Schibler, J., Eds.; Verlag Marie Leidorf: Rahden/Westf, Germany, 1999; pp. 285–300. [Google Scholar]
- Simpson, G.G. Large pleistocene felines of North America. Am. Mus. Novit. 1941, 1136, 1–27. [Google Scholar]
- Boudadi-Maligne, M.; Escarguel, G. A biometric re-evaluation of recent claims for early upper palaeolithic wolf domestication in Eurasia. J. Archaeol. Sci. 2014, 45, 80–89. [Google Scholar] [CrossRef]
- Escarguel, G. Macroécologie en Temps Profond: Motifs, Rythmes et Modalities des Changements de Biodiversité à L’échelle des Temps Géologiques; Université Claude Bernard Lyon 1: Lyon, France, 2008. [Google Scholar]
- Brayard, A.; Escarguel, G.; Bucher, H. The biogeography of early triassic ammonoid faunas: Clusters, gradients, and networks. Geobios 2007, 40, 749–765. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Capitan, A.; Pausch, H.; Stothard, P.; van Binsbergen, R.; Brondum, R.F.; Liao, X.; Djari, A.; Rodriguez, S.C.; Grohs, C.; et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat. Genet. 2014, 46, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hanotte, O.; Mwai, O.A.; Dessie, T.; Bashir, S.; Diallo, B.; Agaba, M.; Kim, K.; Kwak, W.; Sung, S.; et al. The genome landscape of indigenous African cattle. Genome Biol. 2017, 18, 34. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qin, X.; Song, X.Z.; Jiang, H.; Shen, Y.; Durbin, K.J.; Lien, S.; Kent, M.P.; Sodeland, M.; Ren, Y.; et al. Bos taurus genome assembly. BMC Genom. 2009, 10, 180. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Sempere, G.; Moazami-Goudarzi, K.; Eggen, A.; Laloe, D.; Gautier, M.; Flori, L. WIDDE: A web-interfaced next generation database for genetic diversity exploration, with a first application in cattle. BMC Genom. 2015, 16, 940. [Google Scholar] [CrossRef] [PubMed]
- Matukumalli, L.K.; Lawley, C.T.; Schnabel, R.D.; Taylor, J.F.; Allan, M.F.; Heaton, M.P.; O’Connell, J.; Moore, S.S.; Smith, T.P.; Sonstegard, T.S.; et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS ONE 2009, 4, e5350. [Google Scholar] [CrossRef] [PubMed]
- Patterson, N.; Price, A.L.; Reich, D. Population structure and eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef] [PubMed]
- Hobolth, A.; Christensen, O.F.; Mailund, T.; Schierup, M.H. Genomic relationships and speciation times of human, chimpanzee, and gorilla inferred from a coalescent hidden Markov model. PLoS Genet. 2007, 3, e7. [Google Scholar] [CrossRef] [PubMed]
- Mailund, T.; Dutheil, J.Y.; Hobolth, A.; Lunter, G.; Schierup, M.H. Estimating divergence time and ancestral effective population size of Bornean and Sumatran orangutan subspecies using a coalescent hidden Markov model. PLoS Genet. 2011, 7, e1001319. [Google Scholar] [CrossRef] [PubMed]
- Dutheil, J.Y.; Gaillard, S.; Stukenbrock, E.H. MafFilter: A highly flexible and extensible multiple genome alignment files processor. BMC Genom. 2014, 15, 53. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.E.; Matukumalli, L.K.; Sonstegard, T.S.; Shade, L.L.; Van Tassell, C.P. Genomic divergences among cattle, dog and human estimated from large-scale alignments of genomic sequences. BMC Genom. 2006, 7, 140. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Turney, C.; Hughen, K.A.; Brook, B.W.; McDonald, H.G.; Bradshaw, C.J. Abrupt warming events drove Late Pleistocene Holarctic megafaunal turnover. Science 2015, 349, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, E.D.; Nogues-Bravo, D.; Orlando, L.; Weinstock, J.; Binladen, J.; Marske, K.A.; Ugan, A.; Borregaard, M.K.; Gilbert, M.T.; Nielsen, R.; et al. Species-specific responses of Late Quaternary megafauna to climate and humans. Nature 2011, 479, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Kirillova, I.V.; Zanina, O.G.; Chernova, O.F.; Lapteva, E.G.; Trofimova, S.S.; Lebedev, V.S.; Tiunov, A.V.; Soares, A.E.R.; Shidlovskiy, F.K.; Shapiro, B. An ancient bison from the mouth of the Rauchua River (Chukotka, Russia). Quat. Res. 2015, 84, 232–245. [Google Scholar] [CrossRef]
- Brugal, J.P.; Croitor, R. Evolution, ecology and biochronology of herbivore associations in Europe during the last 3 million years. Quaternaire 2007, 18, 129–152. [Google Scholar] [CrossRef]
- Maniakas, I.; Kostopoulos, D.S. Morphometric-palaeoecological discrimination between bison populations of the western Palaearctic. Geobios 2017, 50, 155–171. [Google Scholar] [CrossRef]
- Wu, D.-D.; Ding, X.-D.; Wang, S.; Wojcik, J.M.; Zhang, Y.; Tokarska, M.; Li, Y.; Wang, M.-S.; Faruque, O.; Nielsen, R.; et al. Pervasive introgression facilitated domestication and adaptation in the Bos species complex. Nat. Ecol. Evol. 2018, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Park, S.D.; Magee, D.A.; McGettigan, P.A.; Teasdale, M.D.; Edwards, C.J.; Lohan, A.J.; Murphy, A.; Braud, M.; Donoghue, M.T.; Liu, Y.; et al. Genome sequencing of the extinct Eurasian wild aurochs, Bos primigenius, illuminates the phylogeography and evolution of cattle. Genome Biol. 2015, 16, 234. [Google Scholar] [CrossRef] [PubMed]
- Achilli, A.; Olivieri, A.; Pellecchia, M.; Uboldi, C.; Colli, L.; Al-Zahery, N.; Accetturo, M.; Pala, M.; Kashani, B.H.; Perego, U.A.; et al. Mitochondrial genomes of extinct aurochs survive in domestic cattle. Curr. Biol. 2008, 18, R157–R158. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.K.; Pritchard, J.K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, R.A.; Taylor, J.F.; Van Tassell, C.P.; Barendse, W.; Eversole, K.A.; Gill, C.A.; Green, R.D.; Hamernik, D.L.; Kappes, S.M.; Lien, S.; et al. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 2009, 324, 528–532. [Google Scholar] [PubMed]
- Patterson, N.; Moorjani, P.; Luo, Y.; Mallick, S.; Rohland, N.; Zhan, Y.; Genschoreck, T.; Webster, T.; Reich, D. Ancient admixture in human history. Genetics 2012, 192, 1065–1093. [Google Scholar] [CrossRef] [PubMed]
- Champlot, S.; Berthelot, C.; Pruvost, M.; Bennett, E.A.; Grange, T.; Geigl, E.M. An efficient multistrategy DNA decontamination procedure of PCR reagents for hypersensitive PCR applications. PLoS ONE 2010, 5, e13042. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Sawyer, S.; Meyer, M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2012, 40, e3. [Google Scholar] [CrossRef] [PubMed]
- Pruvost, M.; Grange, T.; Geigl, E.-M. Minimizing DNA contamination by using UNG-coupled quantitative real-time PCR on degraded DNA samples: Application to ancient DNA studies. Biotechniques 2005, 38, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Poinar, H.N. Ancient DNA: Do it right or not at all. Science 2000, 289, 1139. [Google Scholar] [CrossRef] [PubMed]
- Kahlke, R.D.; García, N.; Kostopoulos, D.S.; Lacombat, F.; Lister, A.M.; Mazza, P.P.A.; Spassov, N.; Titov, V.V. Western Palaearctic palaeoenvironmental conditions during the Early and early Middle Pleistocene inferred from large mammal communities, and implications for hominin dispersal in Europe. Quat. Sci. Rev. 2011, 30, 1368–1395. [Google Scholar] [CrossRef]
- Spassov, N.; Stoytchev, T. On the origin of the wisent, Bison bonasus (Linnaeus, 1758): Presence of the wisent in the upper Palaeolithic art of Eurasia. In Advances in Vertebrate Paleontology “Hen to Panta”; “Emil Racoviță” Institute of Speleology: Bucharest, Romania, 2003; pp. 125–130. [Google Scholar]
- Pivonka, P. Multiscale Mechanobiology of Bone Remodeling and Adaptation; Springer: Cham, Switzerland, 2018; p. 286. [Google Scholar]
- van Asperen, E.N.; Kahlke, R.D. Dietary traits of the late early Pleistocene Bison menneri (Bovidae, Mammalia) from its type site Untermassfeld (Central Germany) and the problem of Pleistocene ‘wood bison’. Quat. Sci. Rev. 2017, 177, 299–313. [Google Scholar] [CrossRef]
- Bocherens, H.; Hofman-Kaminska, E.; Drucker, D.G.; Schmolcke, U.; Kowalczyk, R. European bison as a refugee species? Evidence from isotopic data on early Holocene bison and other large herbivores in northern Europe. PLoS ONE 2015, 10, e0115090. [Google Scholar] [CrossRef] [PubMed]
- Palombo, M.R. Deconstructing mammal dispersals and faunal dynamics in SW Europe during the Quaternary. Quat. Sci. Rev. 2014, 96, 50–71. [Google Scholar] [CrossRef]
- Markova, A.K.; Puzachenko, A.Y.; van Kolfschoten, T.; Kosintsev, P.A.; Kuznetsova, T.V.; Tikhonov, A.N.; Bachura, O.P.; Ponomarev, D.V.; van der Pflicht, J.; Kuitems, M. Changes in the Eurasian distribution of the musk ox (Ovibos moschatus) and the extinct bison (Bison priscus) during the last 50 ka BP. Quat. Int. 2015, 378, 99–110. [Google Scholar] [CrossRef]
- Simpson, G.G. Principles of Animal Taxonomy; Columbia University Press: New York, NY, USA, 1961. [Google Scholar]
- Van Gelder, R.G. Mammalian Hybrids and Generic Limits. Am. Mus. Novit. 1977, 2635, 1–25. [Google Scholar]
- Lott, D.F. American Bison: A Natural History; University of California Press: Berkeley, CA, USA, 2002. [Google Scholar]
- Post, D.M.; Armbrust, T.S.; Horne, E.A.; Goheen, J.R. Sexual segregation results in differences in content and quality of bison (Bos bison) diets. J. Mammal. 2001, 82, 407–413. [Google Scholar] [CrossRef]
- Mooring, M.S.; Reisig, D.D.; Osborne, E.R.; Kanallakan, A.L.; Hall, B.M.; Schaad, E.W.; Wiseman, D.S.; Huber, R.R. Sexual segregation in bison: A test of multiple hypotheses. Behaviour 2005, 142, 897–927. [Google Scholar] [CrossRef]
- Komers, P.E.; Messier, F.; Gates, C.C. Group structure in wood bison: Nutritional and reproductive determinants. Can. J. Zool. 1993, 71, 1367–1371. [Google Scholar] [CrossRef]
- Krasińska, M.; Krasiński, Z.A. European Bison: The Nature Monograph, 2nd ed.; Springer: Heidelberg, Germany, 2013. [Google Scholar]
- Rohland, N.; Reich, D.; Mallick, S.; Meyer, M.; Green, R.E.; Georgiadis, N.J.; Roca, A.L.; Hofreiter, M. Genomic DNA sequences from mastodon and woolly mammoth reveal deep speciation of forest and savanna elephants. PLoS Biol. 2010, 8, e1000564. [Google Scholar] [CrossRef] [PubMed]
- Schnitzler, A.; Granado, J.; Putelat, O.; Arbogast, R.M.; Drucker, D.; Eberhard, A.; Schmutz, A.; Klaefiger, Y.; Lang, G.; Salzburger, W.; et al. Genetic diversity, genetic structure and diet of ancient and contemporary red deer (Cervus elaphus L.) from north-eastern France. PLoS ONE 2018, 13, e0189278. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age Estimates of the Nodes (kya) | |
| Node | Age [95% HPD] |
| Root Bovina | 1549 [1736–1366] |
| Bos p. taurus/B. bonasus | 1273 [1436–1117] |
| P. grunniens/B. priscus | 536 [608,468] |
| B. priscus | 203 [224,177] |
| Modern B. bison | 14 [17,12] |
| All B. bonasus | 395 [445,343] |
| B. bonasus Bb1 | 214 [240,183] |
| B. bonasus Bb2 | 116 [130,98] |
| Clock Rate Estimates (Per Site * Year) | |
| Partition | Rate [95% HPD] |
| HVR | 3.0 [2.5–3.5] × 10−7 |
| RNA | 1.7 [1.4–1.9] × 10−8 |
| Coding (1st–2d positions) | 1.55 [1.35–1.75] × 10−8 |
| Coding (3d position) | 5.35 [4.75–6.0] × 10−8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grange, T.; Brugal, J.-P.; Flori, L.; Gautier, M.; Uzunidis, A.; Geigl, E.-M. The Evolution and Population Diversity of Bison in Pleistocene and Holocene Eurasia: Sex Matters. Diversity 2018, 10, 65. https://doi.org/10.3390/d10030065
Grange T, Brugal J-P, Flori L, Gautier M, Uzunidis A, Geigl E-M. The Evolution and Population Diversity of Bison in Pleistocene and Holocene Eurasia: Sex Matters. Diversity. 2018; 10(3):65. https://doi.org/10.3390/d10030065
Chicago/Turabian StyleGrange, Thierry, Jean-Philip Brugal, Laurence Flori, Mathieu Gautier, Antigone Uzunidis, and Eva-Maria Geigl. 2018. "The Evolution and Population Diversity of Bison in Pleistocene and Holocene Eurasia: Sex Matters" Diversity 10, no. 3: 65. https://doi.org/10.3390/d10030065
APA StyleGrange, T., Brugal, J.-P., Flori, L., Gautier, M., Uzunidis, A., & Geigl, E.-M. (2018). The Evolution and Population Diversity of Bison in Pleistocene and Holocene Eurasia: Sex Matters. Diversity, 10(3), 65. https://doi.org/10.3390/d10030065