Intraspecific Variation in Commiphora wightii Populations Based on Internal Transcribed Spacer (ITS1-5.8S-ITS2) Sequences of rDNA

Abstract
:1. Introduction
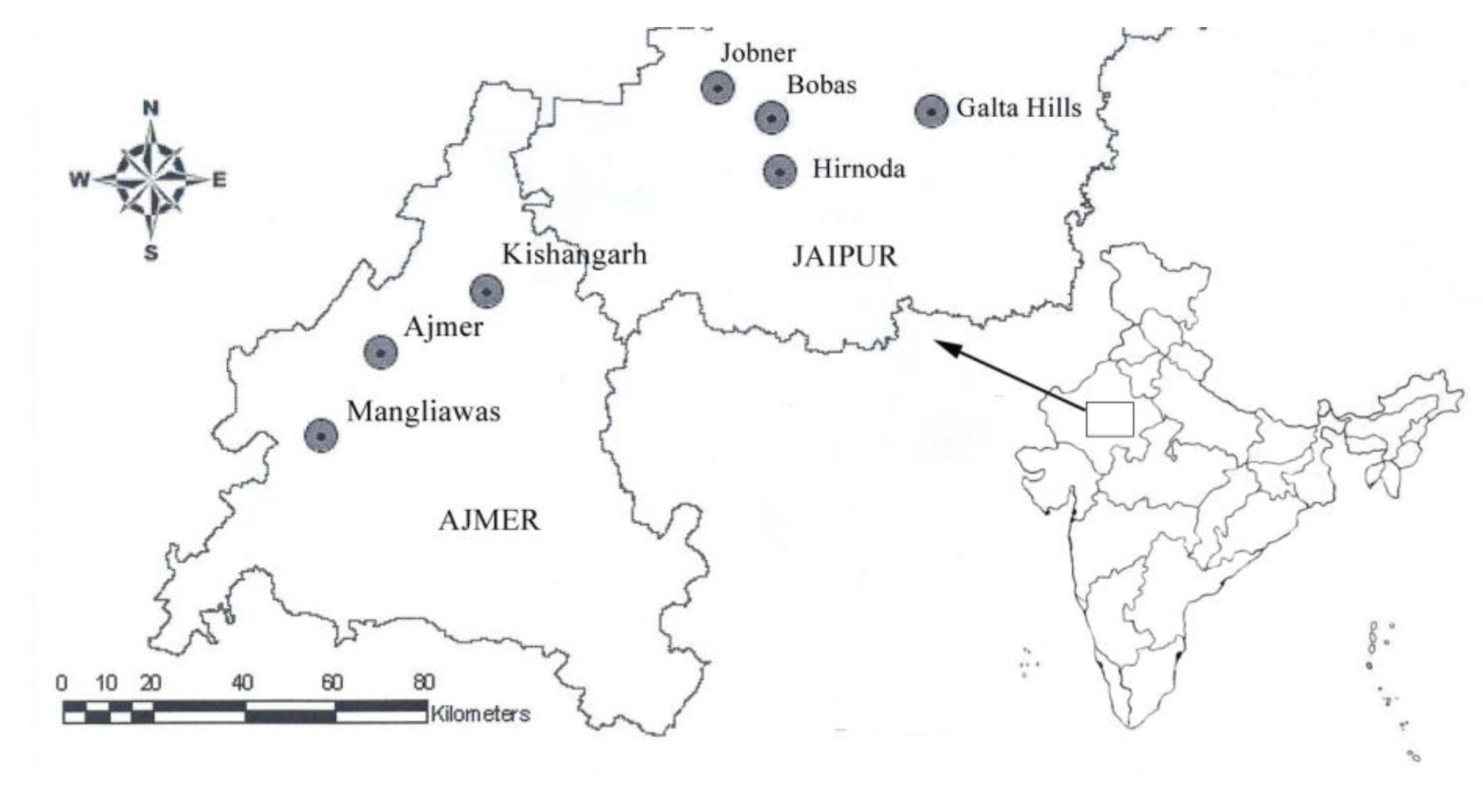
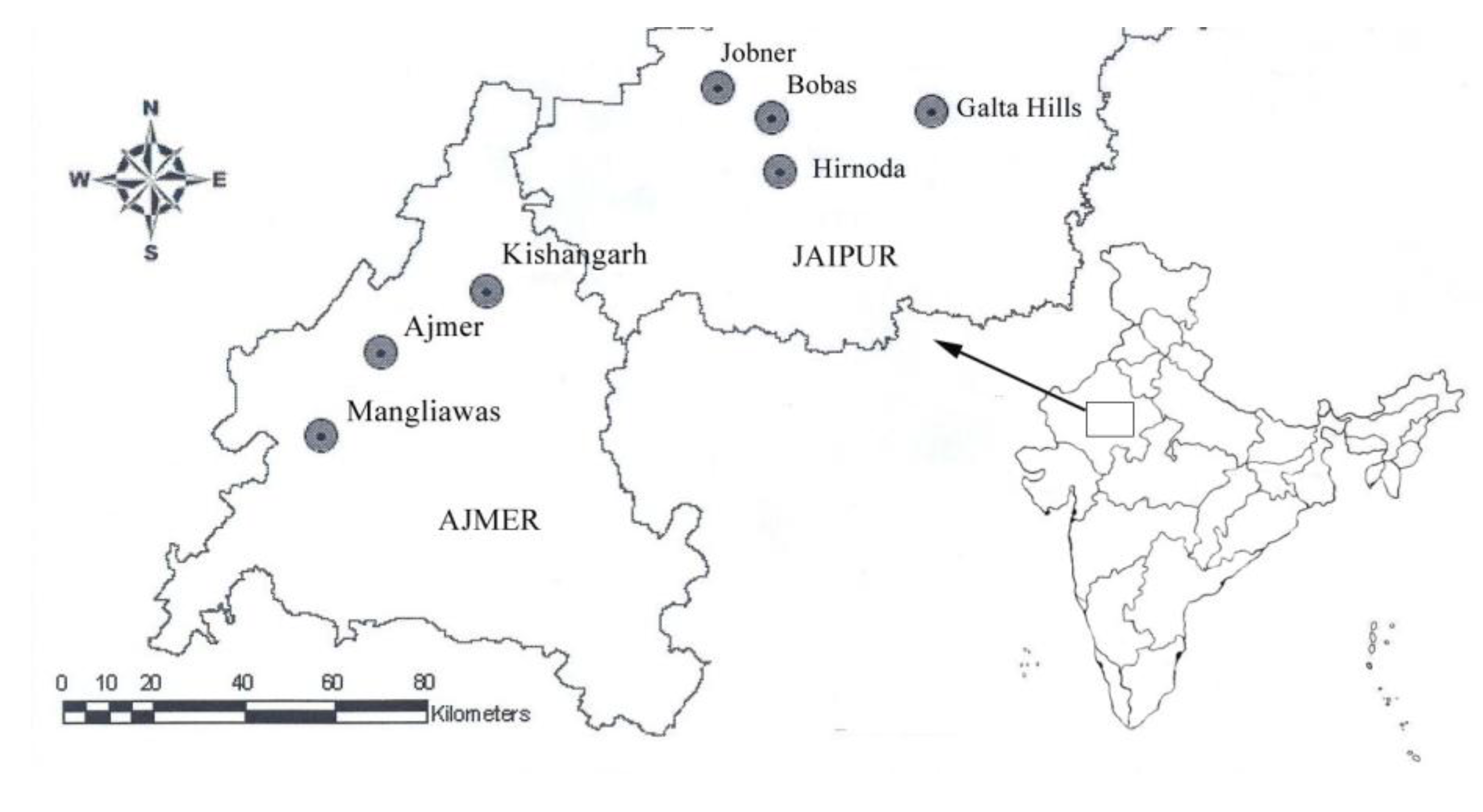
2. Material and Methods

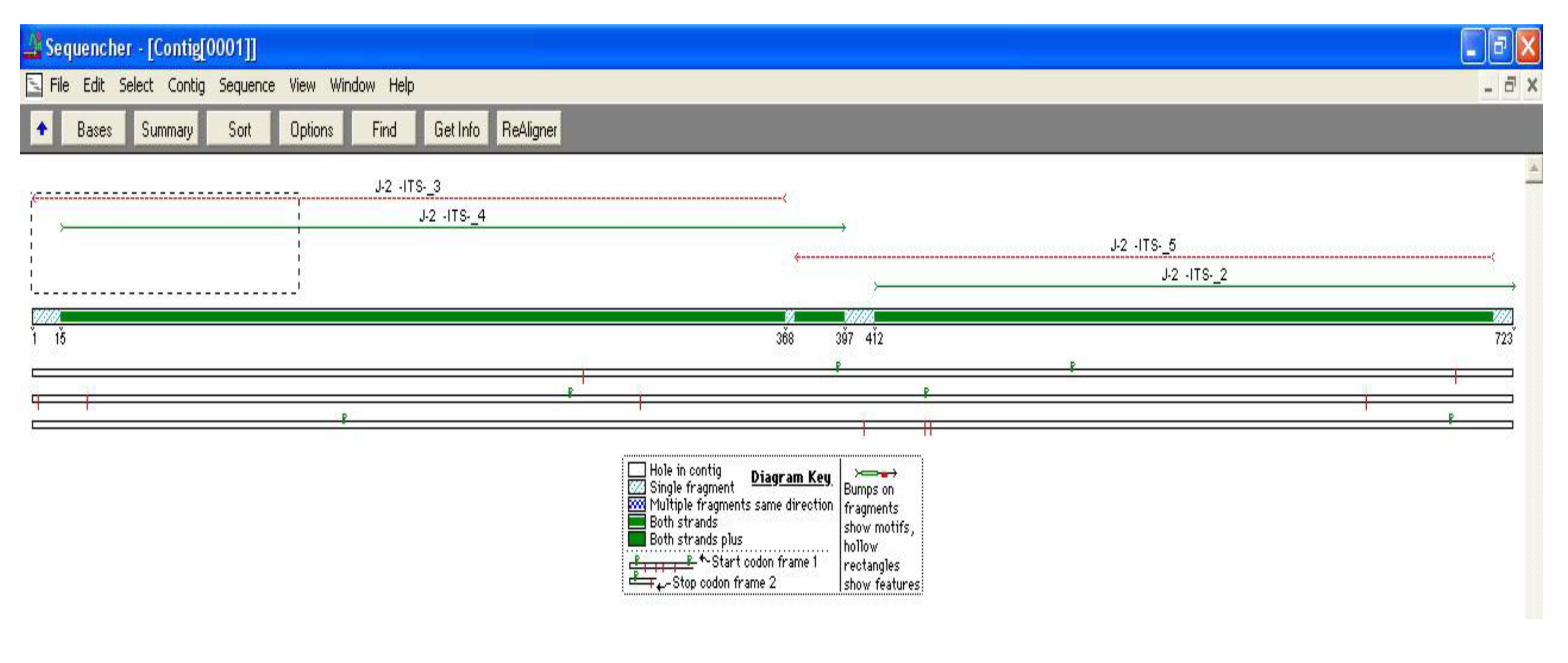
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Terrain | Population Code | Sample size | Latitude | Longitude | Elevation (ft) |
| Mangliawas | Hilly | M | 7 | 26.283 | 74.500 | 1,433 |
| Ajmer | Hilly | A | 4 | 26.450 | 74.633 | 1,548 |
| Kishangarh | Hilly | KG | 4 | 26.566 | 74.866 | 1,420 |
| Jobner | Plains | J | 8 | 26.966 | 75.383 | 1,243 |
| Bobas | Plains | B | 3 | 26.905 | 75.498 | 1,220 |
| Hirnoda | Plains | H | 3 | 26.885 | 75.333 | 1,116 |
| Galta Hills | Hilly | GH | 3 | 26.916 | 75.857 | 1,698 |
2.1. Data Analysis
3. Results and Discussion
| A | T | C | G | |
| A | - | 4.84 | 8.22 | 22.43 |
| T | 3.94 | - | 10.13 | 8.57 |
| C | 3.94 | 5.96 | - | 8.57 |
| G | 10.33 | 4.84 | 4.22 | - |
| Population | π | θw |
|---|---|---|
| Mangliawas | 0.03473 (0.00908) | 0.03446 (0.01590) |
| Ajmer | 0.03078 (0.01080) | 0.03148 (0.01742) |
| Kishangarh | 0.03650 (0.00843) | 0.03424 (0.01890) |
| Jobner | 0.04445 (0.00550) | 0.04966 (0.02180) |
| Bobas | 0.03661 (0.01121) | 0.03614 (0.02210) |
| Hirnoda | 0.01107 (0.00374) | 0.01107 (0.00709) |
| Galta Hills | 0.05493 (0.01707) | 0.05307 (0.03221) |
| All Samples | 0.03905 (0.00307) | 0.05418 (0.01685) |
| Source of variation | d.f. | SSD | Estimated variance | Total variance (%) | ΦST | p-value |
|---|---|---|---|---|---|---|
| Among population | 6 | 301.118 | 6.046 | 21 | 0.206 | 0.01 |
| Within population | 25 | 508.796 | 23.359 | 79 | ||
| Total | 31 | 885.094 | 29.405 |
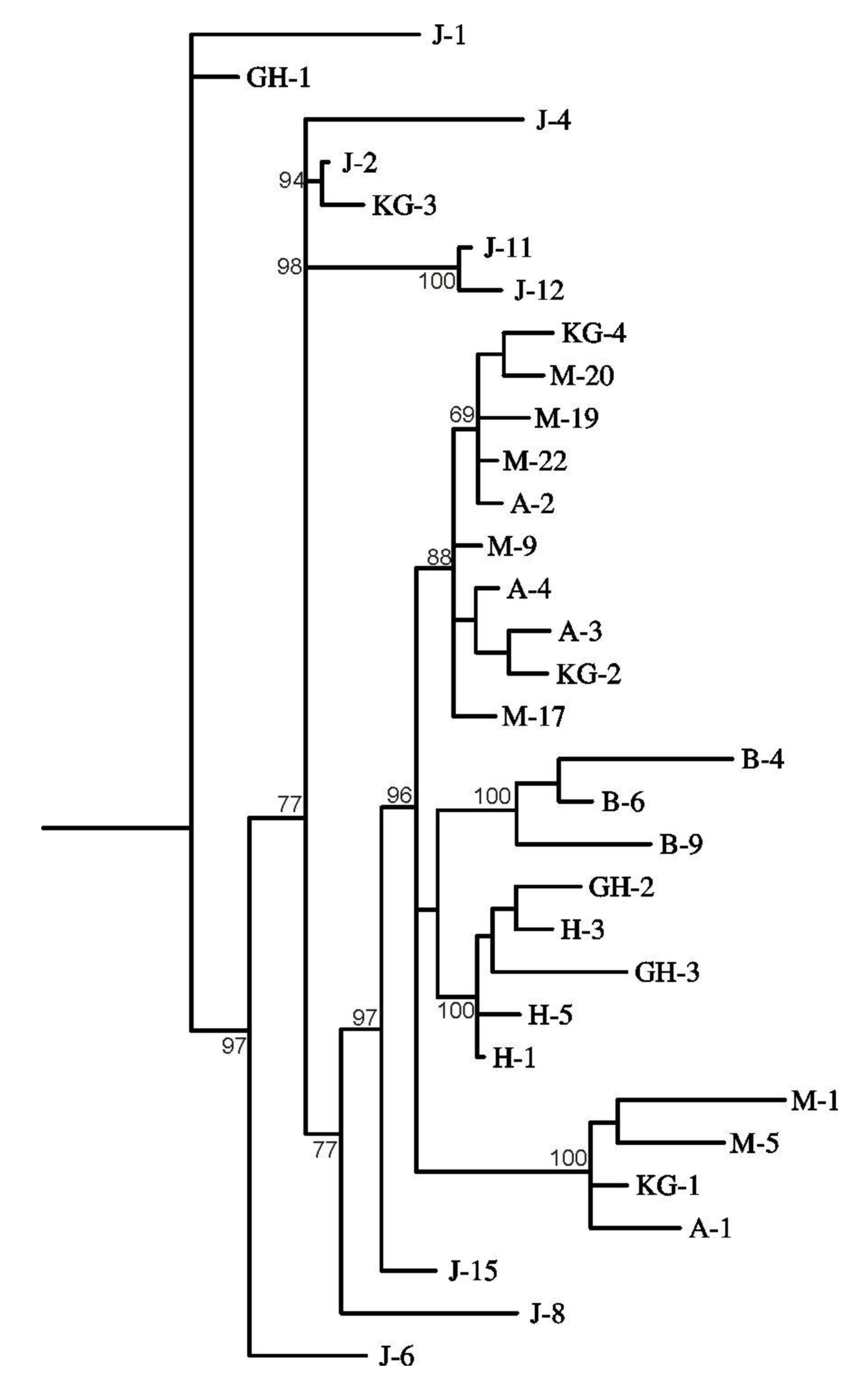
4. Conclusions

Acknowledgements
References and Notes
- APG, II. An update of the Angiosperm Phylogenetic Group classification for the orders and families of flowering plants: APG II. Bot. J. Lin. Soc. 2003, 141, 399–436. [Google Scholar] [CrossRef]
- Gupta, P.; Shivanna, K.R.; Mohan Ram, H.Y. Apomixis and polyembryony in the guggul plant, Commiphora wightii. Ann. Bot. 1996, 78, 67–72. [Google Scholar] [CrossRef]
- Murray, M.T. The Healing Power of Herbs: The Enlightened Person’s Guide to the Wonders of Medicinal Plants; Prima Publishers: Roseville, CA, USA, 1995. [Google Scholar]
- El Ashry, E.S.; Rashed, N.; Salama, O.M.; Saleh, A. Components, therapeutic value and uses of myrrh. Pharmazie 2003, 58, 163–168. [Google Scholar]
- Singh, R.B.; Niaz, M.A.; Ghosh, G. Hypolipidemic and antioxidant effects of Commiphora mukul as an adjunct to dietary therapy in patients with hypercholesterolemia. Cardiovasc. Drugs Ther. 1994, 8, 659–664. [Google Scholar] [CrossRef]
- Deng, R. Therapeutic effects of guggul and its constituent guggulsterone: Cardiovascular benefits. Cardiovasc. Drug Rev. 2007, 25, 375–390. [Google Scholar]
- Sarfaraz, S.; Siddiqui, I.A.; Syed, D.N.; Afaq, F.; Mukhtar, H. Guggulsterone modulates MAPK and NF-kappaB pathways and inhibits skin tumorigenesis in SENCAR mice. Carcinogenesis 2008, 29, 2011–2018. [Google Scholar] [CrossRef]
- Singh, S.V.; Choi, S.; Zeng, Y.; Hahm, E.R.; Xiao, D. Guggulsterone-induced apoptosis in human prostate cancer cells is caused by reactive oxygen intermediate dependent activation of c-Jun NH2-terminal kinase. Cancer Res. 2007, 67, 7439–7449. [Google Scholar] [CrossRef]
- Xiao, D.; Singh, S.V. Z-Guggulsterone, a constituent of Ayurvedic medicinal plant Commiphora mukul, inhibits angiogenesis in vitro and in vivo. Mol. Cancer Ther. 2008, 7, 171–180. [Google Scholar] [CrossRef]
- Khan, T.I.; Dular, A.K.; Deepika, M.S. Biodiversity conservation in the Thar Desert; with emphasis on endemic and medicinal plants. Environmentalist 2003, 23, 137–144. [Google Scholar] [CrossRef]
- Soltis, D.; Soltis, P. Choosing an approach and an appropriate gene for phylogenetic analysis. In Molecular Systematics of Plants II. DNA Sequencing; Soltis, D., Soltis, P., Doyle, J., Eds.; Kluwer Academic Publishers: Boston, MA, USA, 1998; pp. 1–42. [Google Scholar]
- Alvarez, I.; Wendel, J.F. Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phylogenet. Evol. 2003, 29, 417–434. [Google Scholar] [CrossRef]
- Baldwin, B.G.; Markos, S. Phylogenetic utility of the external transcribed spacer (ETS) of 18S-26S rDNA: Congruence of ETS and ITS trees of Calycadenia (Compositae). Mol. Phylogenet. Evol. 1998, 10, 449–463. [Google Scholar] [CrossRef]
- Baldwin, B.; Sanderson, M.; Porter, J.; Wojciechowski, M.; Campbell, C.; Donoghue, M. The ITS region of nuclear ribosomal DNA- a valuable source of evidence on angiosperm phylogeny. Ann. Mo. Bot. Gard. 1995, 82, 247–277. [Google Scholar] [CrossRef]
- Hershkovitz, M.A.; Zimmer, E.A.; Hahn, W.J. Ribosomal DNA sequences and angiosperm systematics. In Molecular Systematics and Plant Evolution; Hollingsworth, P.M., Bateman, R.M., Gornall, R.J., Eds.; Taylor & Francis: London, UK, 1999; pp. 268–326. [Google Scholar]
- Kelch, D.G.; Baldwin, B.G. Phylogeny and ecological radiation of New World thistles (Cirsium, Cardueae-Compositae) based on ITS and ETS rDNA sequence data. Mol. Ecol. 2003, 12, 141–151. [Google Scholar] [CrossRef]
- Lee, J.Y.; Mummenho, V.K.; Bowman, J.L. Allopolyploidization and evolution of species with reduced floral structures in Lepidium L. (Brassicaceae). Proc. Natl. Acad. Sci. USA 2002, 99, 16835–16840. [Google Scholar] [CrossRef]
- Ainouche, M.L.; Bayer, R.J. On the origins of the tetraploid Bromus species (section Bromus, Poaceae): insights from the internal transcribed spacer sequences of nuclear ribosomal DNA. Genome 1997, 40, 730–743. [Google Scholar] [CrossRef]
- Campbell, C.S.; Wojciechowski, M.F.; Baldwin, B.G.; Alice, L.A.; Donoghue, M.J. Persistent nuclear ribosomal DNA sequence polymorphism in the Amelanchier agamic complex (Rosaceae). Mol. Biol. Evol. 1997, 14, 81–90. [Google Scholar]
- Hughes, C.E.; Bailey, C.D.; Harris, S.A. Divergent and reticulate species relationships in Leucaena (Fabaceae) inferred from multiple data sources: Insights into polyploid origins and nrDNA polymorphism. Am. J. Bot. 2002, 89, 1057–1073. [Google Scholar] [CrossRef]
- Feliner, G.N.; Larena, B.G.; Aguilar, J.F. Fine scale geographic structure, intra-individual polymorphism and recombination in nuclear ribosomal internal transcribed spacers in Armeria (Plumbaginaceae). Ann. Bot. 2004, 93, 189–200. [Google Scholar] [CrossRef]
- Buckler, E.S.; Ippolito, A.; Holtsford, T.P. The evolution of ribosomal DNA: divergent paralogues and phylogenetic implications. Genetics 1997, 145, 821–832. [Google Scholar]
- Becerra, J.X. Insects on plants: macroevolutionary chemical trends in host use. Science 1999, 276, 253–256. [Google Scholar] [CrossRef]
- Becerra, J.X.; Venable, D.L. Nuclear ribosomal DNA phylogeny and its implications for evolutionary trends in Mexican Bursera (Burseraceae). Am. J. Bot. 1999, 86, 1047–1057. [Google Scholar] [CrossRef]
- Becerra, J.X. Evolution of Mexican Bursera (Burseraceae) inferred from ITS, ETS, and 5S nuclear ribosomal DNA sequences. Mol. Phylogenet. Evol. 2003, 26, 300–309. [Google Scholar] [CrossRef]
- Weeks, A.; Daly, D.C.; Simpson, B.B. The phylogenetic history and biogeography of the frankincense and myrrh family (Burseraceae) based on nuclear and chloroplast sequence data. Mol. Phylogenet. Evol. 2005, 35, 85–101. [Google Scholar] [CrossRef]
- Haque, I.; Bandopadhyay, R.; Mukhopadhyay, K. Population genetic structure of the endangered and endemic medicinal plant Commiphora wightii. Mol. Biol. Rep. 2009. [Google Scholar] [CrossRef]
- Milligan, B.G.; Leebens-Mack, J.; Strand, A.E. Conservation genetics: beyond the maintenance of marker diversity. Mol. Ecol. 1994, 3, 423–435. [Google Scholar] [CrossRef]
- Haque, I.; Bandopadhyay, R.; Mukhopadhyay, K. An optimised protocol for fast genomic DNA isolation from high secondary metabolites and gum containing plants. Asian J. Plant Sci. 2008, 7, 304–308. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplifcation and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M., Gelfand, D., Sninsky, J., White, T., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, G.D. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 24, 4876–4882. [Google Scholar]
- Watterson, G.A. On the number of segregating sites in genetical models without recombination. Theor. Pop. Biol. 1975, 7, 256–276. [Google Scholar]
- Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 1978, 89, 583–590. [Google Scholar]
- Rozas, J.; Rozas, R. DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 1999, 15, 174–175. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel, population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967, 27, 209–220. [Google Scholar]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MrBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Posada, D. Selection of Models of DNA Evolution with jModelTest. In Bioinformatics for DNA Sequence Analysis; Posada, D., Ed.; SpringerLink: Secaucus, NJ, USA, 2009; Volume 537, pp. 93–112. [Google Scholar]
- Dover, G.A.; Linares, A.R.; Bowen, T.; Hancock, J.M. Detection and quantification of concerted evolution and molecular drive. Methods Enzymol. 1993, 224, 525–541. [Google Scholar] [CrossRef]
- Appels, R.; Honeycutt, R.L. rDNA: Evolution over a billion years. In DNA systematics; Dutton, S., Ed.; CRC Press: Boca Raton, FL, USA, 1986; pp. 81–135. [Google Scholar]
- Adolfsson, S.; Bengtsson, B.O. The spread of apomixis and its effect on resident genetic variation. J. Evol. Biol. 2007, 20, 1933–1940. [Google Scholar] [CrossRef]
- Nei, M.; Li, W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Haque, I.; Bandopadhyay, R.; Mukhopadhyay, K. Intraspecific Variation in Commiphora wightii Populations Based on Internal Transcribed Spacer (ITS1-5.8S-ITS2) Sequences of rDNA. Diversity 2009, 1, 89-101. https://doi.org/10.3390/d1020089
Haque I, Bandopadhyay R, Mukhopadhyay K. Intraspecific Variation in Commiphora wightii Populations Based on Internal Transcribed Spacer (ITS1-5.8S-ITS2) Sequences of rDNA. Diversity. 2009; 1(2):89-101. https://doi.org/10.3390/d1020089
Chicago/Turabian StyleHaque, Inamul, Rajib Bandopadhyay, and Kunal Mukhopadhyay. 2009. "Intraspecific Variation in Commiphora wightii Populations Based on Internal Transcribed Spacer (ITS1-5.8S-ITS2) Sequences of rDNA" Diversity 1, no. 2: 89-101. https://doi.org/10.3390/d1020089
APA StyleHaque, I., Bandopadhyay, R., & Mukhopadhyay, K. (2009). Intraspecific Variation in Commiphora wightii Populations Based on Internal Transcribed Spacer (ITS1-5.8S-ITS2) Sequences of rDNA. Diversity, 1(2), 89-101. https://doi.org/10.3390/d1020089