
Synthesis of N,N-Dimethylaminopropyl Derivative of A Blood Sugar Antigen



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
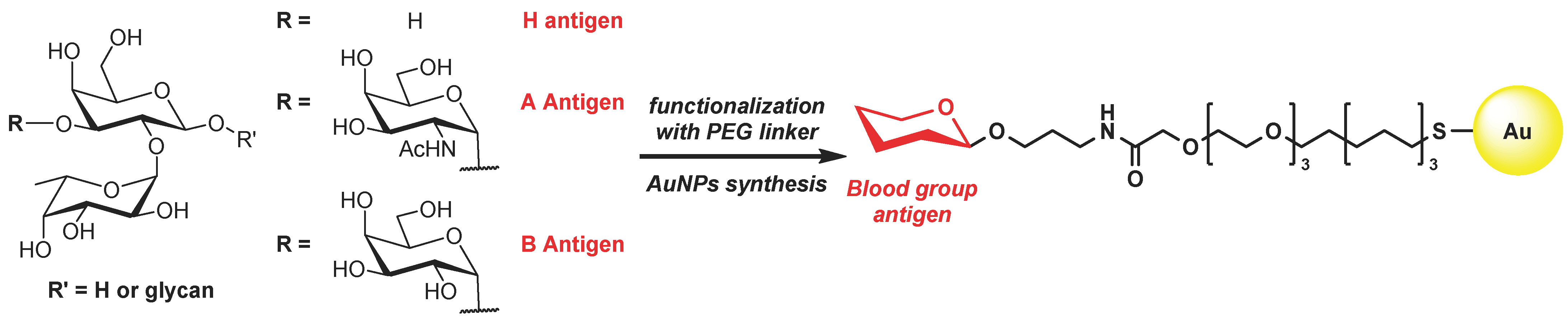
1. Introduction
2. Results and Discussion
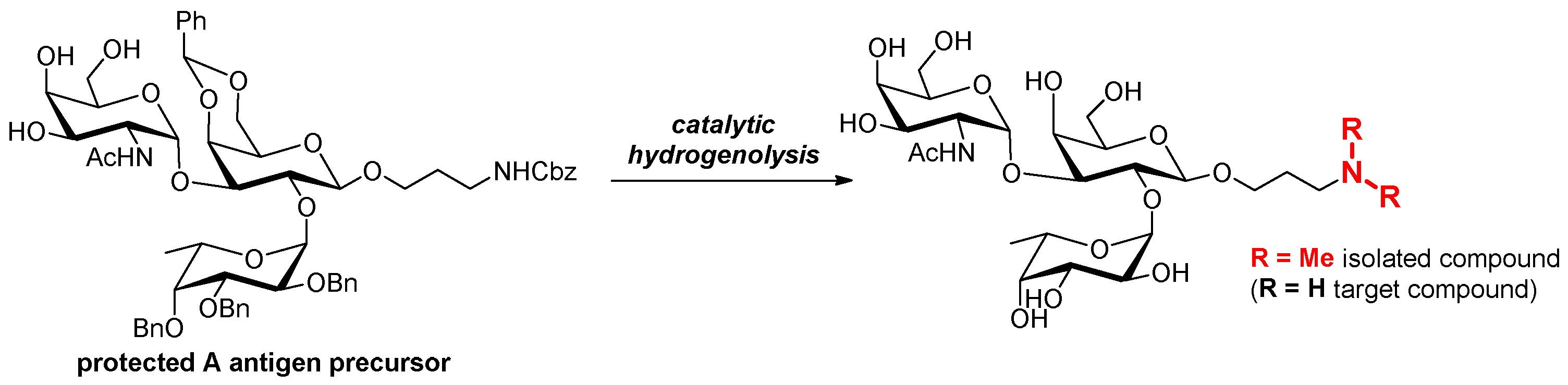
2.1. Retrosynthesis
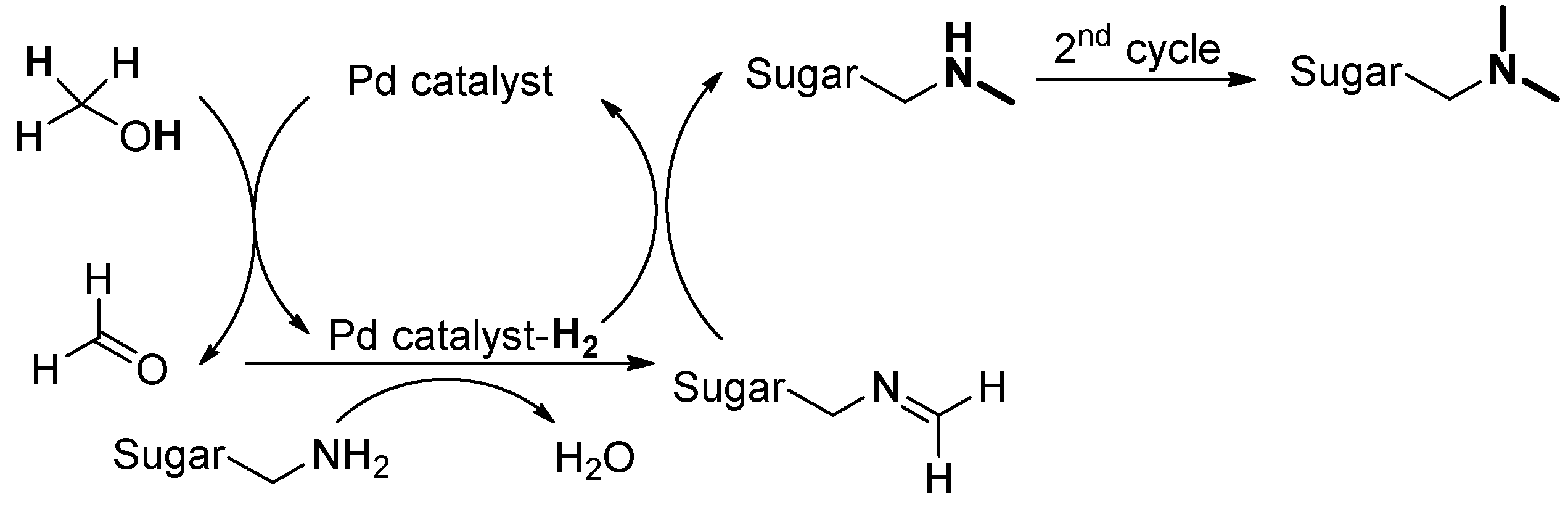
2.2. Synthesis of Trisaccharide Antigen A Derivative 5
3. Materials and Methods
3.1. General Remarks
3.2. Synthetic Procedures and Characterizations
3.2.1. N-(Benzyloxycarbonyl)aminopropyl-3-O-acetyl-4,6-O-benzylidene-β-d-galactopyranoside (2)
3.2.2. N-(Benzyloxycarbonyl)aminopropyl-3-O-acetyl-4,6-O-benzylidene-2-O-(2′,3′,4′-tri-O-benzyl-α-l-fucopyranosyl)-β-d-galactopyranoside (4)
3.2.3. N-(Benzyloxycarbonyl)aminopropyl-4,6-O-benzylidene-2-O-(2′,3′,4′-tri-O-benzyl-α-l-fucopyranosyl)-β-d-galactopyranoside (5)
3.2.4. N-(Benzyloxycarbonyl)aminopropyl-2-O-(2′,3′,4′-tri-O-benzyl-α-l-fucopyranosyl)-3-O-(3″,4″,6″-tri-O-acetyl-2-azido-2-deoxy-α-d-galactopyranosyl)-4,6-O-benzylidene-β-d-galactopyranoside (7)
3.2.5. N-(Benzyloxycarbonyl)aminopropyl-2-O-(2′,3′,4′-tri-O-benzyl-α-l-fucopyranosyl)-3-O-(2-azido-2-deoxy-α-d-galactopyranosyl)-4,6-O-benzylidene-β-d-galactopyranoside (8)
3.2.6. N-(Benzyloxycarbonyl)aminopropyl-2-O-(2′,3′,4′-tri-O-benzyl-α-l-fucopyranosyl)-3-O-(2-N-acetamido-α-d-galactopyranosyl)-4,6-O-benzylidene-β-d-galactopyranoside (10)
3.2.7. N,N-Dimethylaminopropyl-[2-O-(α-L-fucopyranosyl)-3-O-(2-deoxy-2-acetamido-α-D-galactopyranosyl)]-β-D-galactopyranoside 11
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baram-Pinto, D.; Shukla, S.; Gedanken, A.; Sarid, R. Inhibition of HSV-1 Attachment, Entry, and Cell-to-Cell Spread by Functionalized Multivalent Gold Nanoparticles. Small 2010, 6, 1044–1050. [Google Scholar] [CrossRef]
- Yeh, Y.-C.; Creran, B.; Rotello, V.M. Gold Nanoparticles: Preparation, Properties, and Applications in Bionanotechnology. Nanoscale 2012, 4, 1871–1880. [Google Scholar] [CrossRef]
- Saha, K.; Agasti, S.S.; Kim, C.; Li, X.; Rotello, V.M. Gold Nanoparticles in Chemical and Biological Sensing. Chem. Rev. 2012, 112, 2739–2779. [Google Scholar] [CrossRef] [PubMed]
- Kadhim, R.J.; Karsh, E.H.; Taqi, Z.J.; Jabir, M.S. Biocompatibility of Gold Nanoparticles: In-Vitro and In-Vivo Study. Mater. Today Proc. 2021, 42, 3041–3045. [Google Scholar] [CrossRef]
- Liz-Marzán, L.M. Gold Nanoparticle Research before and after the Brust–Schiffrin Method. Chem. Commun. 2013, 49, 16–18. [Google Scholar] [CrossRef]
- Reimers, J.R.; Ford, M.J.; Halder, A.; Ulstrup, J.; Hush, N.S. Gold Surfaces and Nanoparticles Are Protected by Au(0)–Thiyl Species and Are Destroyed When Au(I)–Thiolates Form. Proc. Natl. Acad. Sci. USA 2016, 113, E1424–E1433. [Google Scholar] [CrossRef]
- Gunawan, C.; Lim, M.; Marquis, C.P.; Amal, R. Nanoparticle–Protein Corona Complexes Govern the Biological Fates and Functions of Nanoparticles. J. Mater. Chem. B 2014, 2, 2060. [Google Scholar] [CrossRef]
- Winzen, S.; Schoettler, S.; Baier, G.; Rosenauer, C.; Mailaender, V.; Landfester, K.; Mohr, K. Complementary Analysis of the Hard and Soft Protein Corona: Sample Preparation Critically Effects Corona Composition. Nanoscale 2015, 7, 2992–3001. [Google Scholar] [CrossRef]
- Compostella, F.; Pitirollo, O.; Silvestri, A.; Polito, L. Glyco-Gold Nanoparticles: Synthesis and Applications. Beilstein J. Org. Chem. 2017, 13, 1008–1021. [Google Scholar] [CrossRef]
- García, I.; Sánchez-Iglesias, A.; Henriksen-Lacey, M.; Grzelczak, M.; Penadés, S.; Liz-Marzán, L.M. Glycans as Biofunctional Ligands for Gold Nanorods: Stability and Targeting in Protein-Rich Media. J. Am. Chem. Soc. 2015, 137, 3686–3692. [Google Scholar] [CrossRef]
- de la Fuente, J.M.; Barrientos, A.G.; Rojas, T.C.; Rojo, J.; Cañada, J.; Fernández, A.; Penadés, S. Gold Glyconanoparticles as Water-Soluble Polyvalent Models to Study Carbohydrate Interactions. Angew. Chem. Int. Ed. 2001, 40, 2257–2261. [Google Scholar] [CrossRef]
- Marradi, M.; García, I.; Penadés, S. Carbohydrate-Based Nanoparticles for Potential Applications in Medicine. Prog. Mol. Biol. Transl. Sci. 2011, 104, 141–173. [Google Scholar]
- Matassini, C.; Marradi, M.; Cardona, F.; Parmeggiani, C.; Robina, I.; Moreno-Vargas, A.J.; Penadés, S.; Goti, A. Gold Nanoparticles Are Suitable Cores for Building Tunable Iminosugar Multivalency. RSC Adv. 2015, 5, 95817–95822. [Google Scholar] [CrossRef]
- Marradi, M.; Chiodo, F.; García, I. Glyconanotechnology and Disease: Gold Nanoparticles Coated with Glycosides as Multivalent Systems for Potential Applications in Diagnostics and Therapy. In Carbohydrates in Drug Design and Discovery; The Royal Society of Chemistry: London, UK, 2015; pp. 89–131. [Google Scholar]
- Mateu Ferrando, R.; Lay, L.; Polito, L. Gold Nanoparticle-Based Platforms for Vaccine Development. Drug Discov. Today Technol. 2020, 38, 57–67. [Google Scholar] [CrossRef]
- Terán-Navarro, H.; Zeoli, A.; Salines-Cuevas, D.; Marradi, M.; Montoya, N.; Gonzalez-Lopez, E.; Ocejo-Vinyals, J.G.; Dominguez-Esteban, M.; Gutierrez-Baños, J.L.; Campos-Juanatey, F.; et al. Gold Glyconanoparticles Combined with 91–99 Peptide of the Bacterial Toxin, Listeriolysin O, Are Efficient Immunotherapies in Experimental Bladder Tumors. Cancers 2022, 14, 2413. [Google Scholar] [CrossRef]
- D’Orazio, G.; Marradi, M.; La Ferla, B. Dual-Targeting Gold Nanoparticles: Simultaneous Decoration with Ligands for Co-Transporters SGLT-1 and B0AT1. Appl. Sci. 2024, 14, 2248. [Google Scholar] [CrossRef]
- Marradi, M.; Chiodo, F.; García, I.; Penadés, S. Glyconanoparticles as Multifunctional and Multimodal Carbohydrate Systems. Chem. Soc. Rev. 2013, 42, 4728. [Google Scholar] [CrossRef]
- Kuroda, S.; Kobashi, Y.; Oi, T.; Amada, H.; Okumura-Kitajima, L.; Io, F.; Yamamto, K.; Kakinuma, H. Discovery of a Potent, Low-Absorbable Sodium-Dependent Glucose Cotransporter 1 (SGLT1) Inhibitor (TP0438836) for the Treatment of Type 2 Diabetes. Bioorg. Med. Chem. Lett. 2018, 28, 3534–3539. [Google Scholar] [CrossRef]
- Deswal, N.; Takkar, P.; Kaur, L.; Ojha, H.; Kumar, R. Synthesis and Bio-Evaluation of Newer Dihydropyridines and Tetrahydropyridines Based Glycomimetic Azasugars. Bioorganic Chem. 2024, 145, 107224. [Google Scholar] [CrossRef]
- Decroocq, C.; Rodríguez-Lucena, D.; Russo, V.; Barragán, T.M.; Mellet, C.O.; Compain, P. The Multivalent Effect in Glycosidase Inhibition: Probing the Influence of Architectural Parameters with Cyclodextrin-based Iminosugar Click Clusters. Chem.-A Eur. J. 2011, 17, 13825–13831. [Google Scholar] [CrossRef]
- D’Orazio, G.; Martorana, A.M.; Filippi, G.; Polissi, A.; De Gioia, L.; La Ferla, B. N-Spirofused Bicyclic Derivatives of 1-Deoxynojirimycin: Synthesis and Preliminary Biological Evaluation. ChemistrySelect 2016, 1, 2444–2447. [Google Scholar] [CrossRef]
- Thépaut, M.; Luczkowiak, J.; Vivès, C.; Labiod, N.; Bally, I.; Lasala, F.; Grimoire, Y.; Fenel, D.; Sattin, S.; Thielens, N.; et al. DC/L-SIGN Recognition of Spike Glycoprotein Promotes SARS-CoV-2 Trans-Infection and Can Be Inhibited by a Glycomimetic Antagonist. PLoS Pathog. 2021, 17, e1009576. [Google Scholar] [CrossRef]
- Sommer, R.; Wagner, S.; Rox, K.; Varrot, A.; Hauck, D.; Wamhoff, E.-C.; Schreiber, J.; Ryckmans, T.; Brunner, T.; Rademacher, C.; et al. Glycomimetic, Orally Bioavailable LecB Inhibitors Block Biofilm Formation of Pseudomonas aeruginosa. J. Am. Chem. Soc. 2018, 140, 2537–2545. [Google Scholar] [CrossRef]
- Antonini, G.; Bernardi, A.; Gillon, E.; Dal Corso, A.; Civera, M.; Belvisi, L.; Varrot, A.; Mazzotta, S. Achieving High Affinity for a Bacterial Lectin with Reversible Covalent Ligands. J. Med. Chem. 2024, 67, 19546–19560. [Google Scholar] [CrossRef]
- Cecioni, S.; Imberty, A.; Vidal, S. Glycomimetics versus Multivalent Glycoconjugates for the Design of High Affinity Lectin Ligands. Chem. Rev. 2015, 115, 525–561. [Google Scholar] [CrossRef]
- Rumio, C.; Dusio, G.; Cardani, D.; La Ferla, B.; D’Orazio, G. Anti-Inflammatory Effects of SGLT1 Synthetic Ligand in In Vitro and In Vivo Models of Lung Diseases. Immuno 2024, 4, 502–520. [Google Scholar] [CrossRef]
- D’Orazio, G.; La Ferla, B. Synthesis of a Small Library of Glycoderivative Putative Ligands of SGLT1 and Preliminary Biological Evaluation. Molecules 2024, 29, 5067. [Google Scholar] [CrossRef]
- D’Orazio, G.; Parisi, G.; Policano, C.; Mechelli, R.; Codacci Pisanelli, G.; Pitaro, M.; Ristori, G.; Salvetti, M.; Nicotra, F.; La Ferla, B. Arsenical C-Glucoside Derivatives with Promising Antitumor Activity. Eur. J. Org. Chem. 2015, 2015, 4620–4623. [Google Scholar] [CrossRef]
- Paiotta, A.; D’Orazio, G.; Palorini, R.; Ricciardiello, F.; Zoia, L.; Votta, G.; De Gioia, L.; Chiaradonna, F.; La Ferla, B. Design, Synthesis, and Preliminary Biological Evaluation of GlcNAc-6P Analogues for the Modulation of Phosphoacetylglucosamine Mutase 1 (AGM1/PGM3). Eur. J. Org. Chem. 2018, 2018, 1946–1952. [Google Scholar] [CrossRef]
- Büll, C.; Boltje, T.J.; van Dinther, E.A.W.; Peters, T.; de Graaf, A.M.A.; Leusen, J.H.W.; Kreutz, M.; Figdor, C.G.; den Brok, M.H.; Adema, G.J. Targeted Delivery of a Sialic Acid-Blocking Glycomimetic to Cancer Cells Inhibits Metastatic Spread. ACS Nano 2015, 9, 733–745. [Google Scholar] [CrossRef]
- D’Orazio, G.; Colombo, L.; Salmona, M.; La Ferla, B. Synthesis and Preliminary Biological Evaluation of Fluorescent Glycofused Tricyclic Derivatives of Amyloid Β-Peptide Ligands. Eur. J. Org. Chem. 2016, 2016, 1660–1664. [Google Scholar] [CrossRef]
- Ravn, V.; Dabelsteen, E. Tissue Distribution of Histo-blood Group Antigens. Apmis 2000, 108, 1–28. [Google Scholar] [CrossRef]
- Daniels, G. Blood Groups on Red Cells, Platelets and Neutrophils. In Blood and Bone Marrow Pathology; Elsevier: Amsterdam, The Netherlands, 2011; pp. 599–617. [Google Scholar]
- Cheng, H.; Cao, X.; Xian, M.; Fang, L.; Cai, T.B.; Ji, J.J.; Tunac, J.B.; Sun, D.; Wang, P.G. Synthesis and Enzyme-Specific Activation of Carbohydrate−Geldanamycin Conjugates with Potent Anticancer Activity. J. Med. Chem. 2005, 48, 645–652. [Google Scholar] [CrossRef]
- Karki, G.; Mishra, V.N.; Mandal, P.K. An Expeditious Synthesis of Blood-Group Antigens, ABO Histo-Blood Group Type II Antigens and Xenoantigen Oligosaccharides with Amino Type Spacer−arms. Glycoconj. J. 2016, 33, 63–78. [Google Scholar] [CrossRef]
- Wegmann, B.; Schmidt, R.R. Synthesis of the H-Disaccharide (2-O-α-l-Fucopyranosyl-d-Galactose) via the Trichloroacetimidate Method. Carbohydr. Res. 1988, 184, 254–261. [Google Scholar] [CrossRef]
- Crawford, C.J.; Seeberger, P.H. Advances in Glycoside and Oligosaccharide Synthesis. Chem. Soc. Rev. 2023, 52, 7773–7801. [Google Scholar] [CrossRef]
- Schmidt, R.R.; Toepfer, A. Glycosylation with Highly Reactive Glycosyl Donors: Efficiency of the Inverse Procedure. Tetrahedron Lett. 1991, 32, 3353–3356. [Google Scholar] [CrossRef]
- Adinolfi, M.; Iadonisi, A.; Ravidà, A.; Schiattarella, M. Versatile Use of Ytterbium(III) Triflate and Acid Washed Molecular Sieves in the Activation of Glycosyl Trifluoroacetimidate Donors. Assemblage of a Biologically Relevant Tetrasaccharide Sequence of Globo H. J. Org. Chem. 2005, 70, 5316–5319. [Google Scholar] [CrossRef]
- Benoiton, N.L. On the Side-reaction of N-alkylation of Amino Groups during Hydrogenolytic Deprotection in Alcohol-containing Solvents. Int. J. Pept. Protein Res. 1993, 41, 611. [Google Scholar] [CrossRef]
- Filira, F.; Biondi, L.; Gobbo, M.; Rocchi, R. N-Alkylation of Amino Acids during Hydrogenolytic Deprotection. Tetrahedron Lett. 1991, 32, 7463–7464. [Google Scholar] [CrossRef]
- Denjean, A.E.F.; Nova, A.; Balcells, D. Borrowing Hydrogen Mechanism in Amine Alkylation by Single Atom Nickel Catalysts. ACS Catal. 2024, 14, 11332–11342. [Google Scholar] [CrossRef]
- Podyacheva, E.; Afanasyev, O.I.; Vasilyev, D.V.; Chusov, D. Borrowing Hydrogen Amination Reactions: A Complex Analysis of Trends and Correlations of the Various Reaction Parameters. ACS Catal. 2022, 12, 7142–7198. [Google Scholar] [CrossRef]
- Yu, X.; Cui, X.; Jing, H.; Qian, B.; Yuan, H.; Shi, F. Recent Development in Synthesis of N-Methylamines with Amines and Methanol. ChemCatChem 2024, 16, e202400291. [Google Scholar] [CrossRef]
- Laferté, S.; Chan, N.W.C.; Sujino, K.; Lowary, T.L.; Palcic, M.M. Intracellular Inhibition of Blood Group A Glycosyltransferase. Eur. J. Biochem. 2000, 267, 4840–4849. [Google Scholar] [CrossRef]
- Gagnon, S.M.L.; Legg, M.S.G.; Sindhuwinata, N.; Letts, J.A.; Johal, A.R.; Schuman, B.; Borisova, S.N.; Palcic, M.M.; Peters, T.; Evans, S. V High-Resolution Crystal Structures and STD NMR Mapping of Human ABO(H) Blood Group Glycosyltransferases in Complex with Trisaccharide Reaction Products Suggest a Molecular Basis for Product Release. Glycobiology 2017, 27, 966–977. [Google Scholar] [CrossRef]
- Valverde, P.; Delgado, S.; Martínez, J.D.; Vendeville, J.-B.; Malassis, J.; Linclau, B.; Reichardt, N.-C.; Cañada, F.J.; Jiménez-Barbero, J.; Ardá, A. Molecular Insights into DC-SIGN Binding to Self-Antigens: The Interaction with the Blood Group A/B Antigens. ACS Chem. Biol. 2019, 14, 1660–1671. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Marzo, E.; Lay, L.; D’Orazio, G. Synthesis of N,N-Dimethylaminopropyl Derivative of A Blood Sugar Antigen. Molbank 2025, 2025, M1985. https://doi.org/10.3390/M1985
Di Marzo E, Lay L, D’Orazio G. Synthesis of N,N-Dimethylaminopropyl Derivative of A Blood Sugar Antigen. Molbank. 2025; 2025(2):M1985. https://doi.org/10.3390/M1985
Chicago/Turabian StyleDi Marzo, Elena, Luigi Lay, and Giuseppe D’Orazio. 2025. "Synthesis of N,N-Dimethylaminopropyl Derivative of A Blood Sugar Antigen" Molbank 2025, no. 2: M1985. https://doi.org/10.3390/M1985
APA StyleDi Marzo, E., Lay, L., & D’Orazio, G. (2025). Synthesis of N,N-Dimethylaminopropyl Derivative of A Blood Sugar Antigen. Molbank, 2025(2), M1985. https://doi.org/10.3390/M1985