
Bis(3-(((4-methoxybenzyl)oxy)methyl)-5,6-dihydro-1,4-dithiin-2-yl)methanol



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
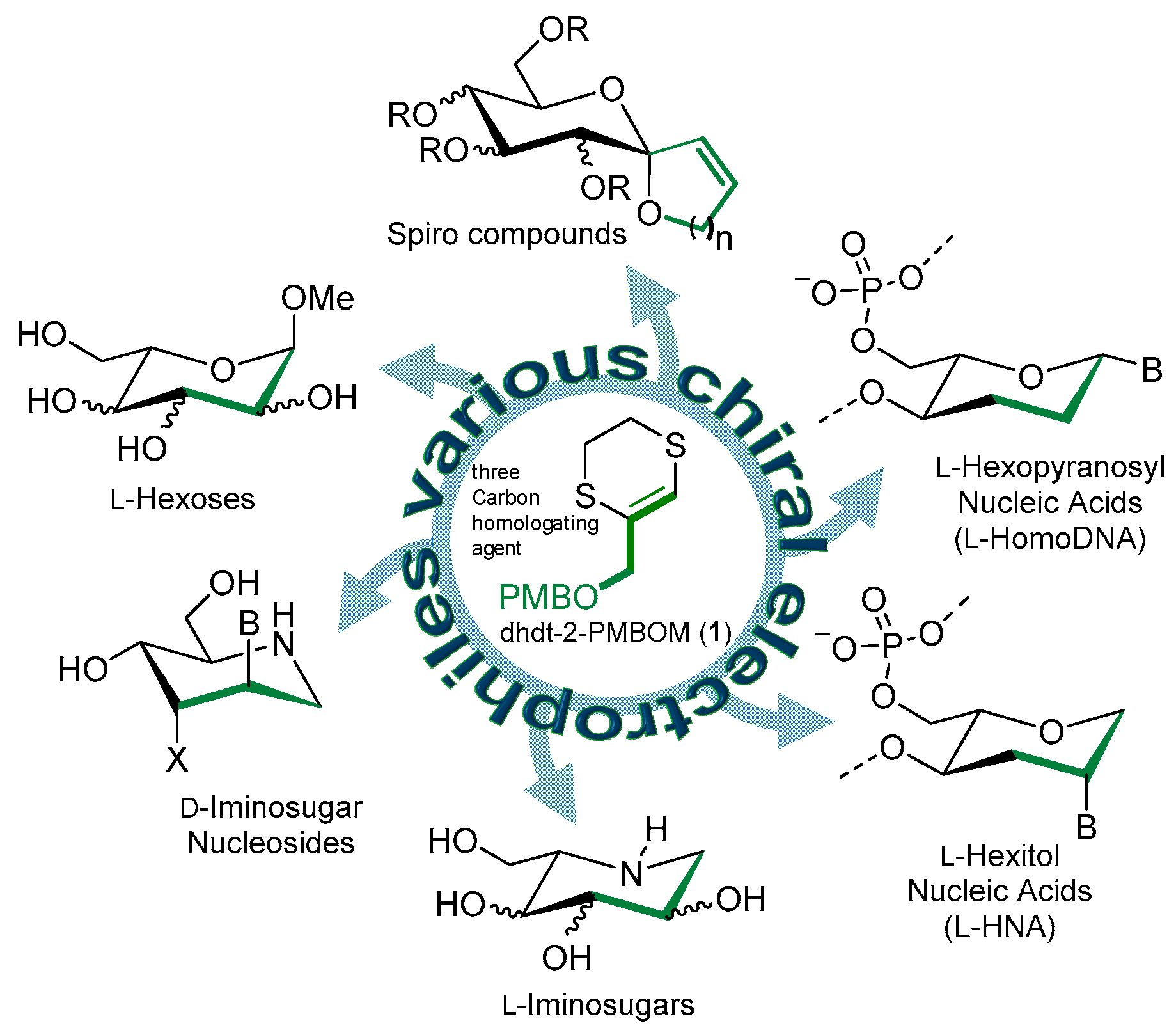
1. Introduction
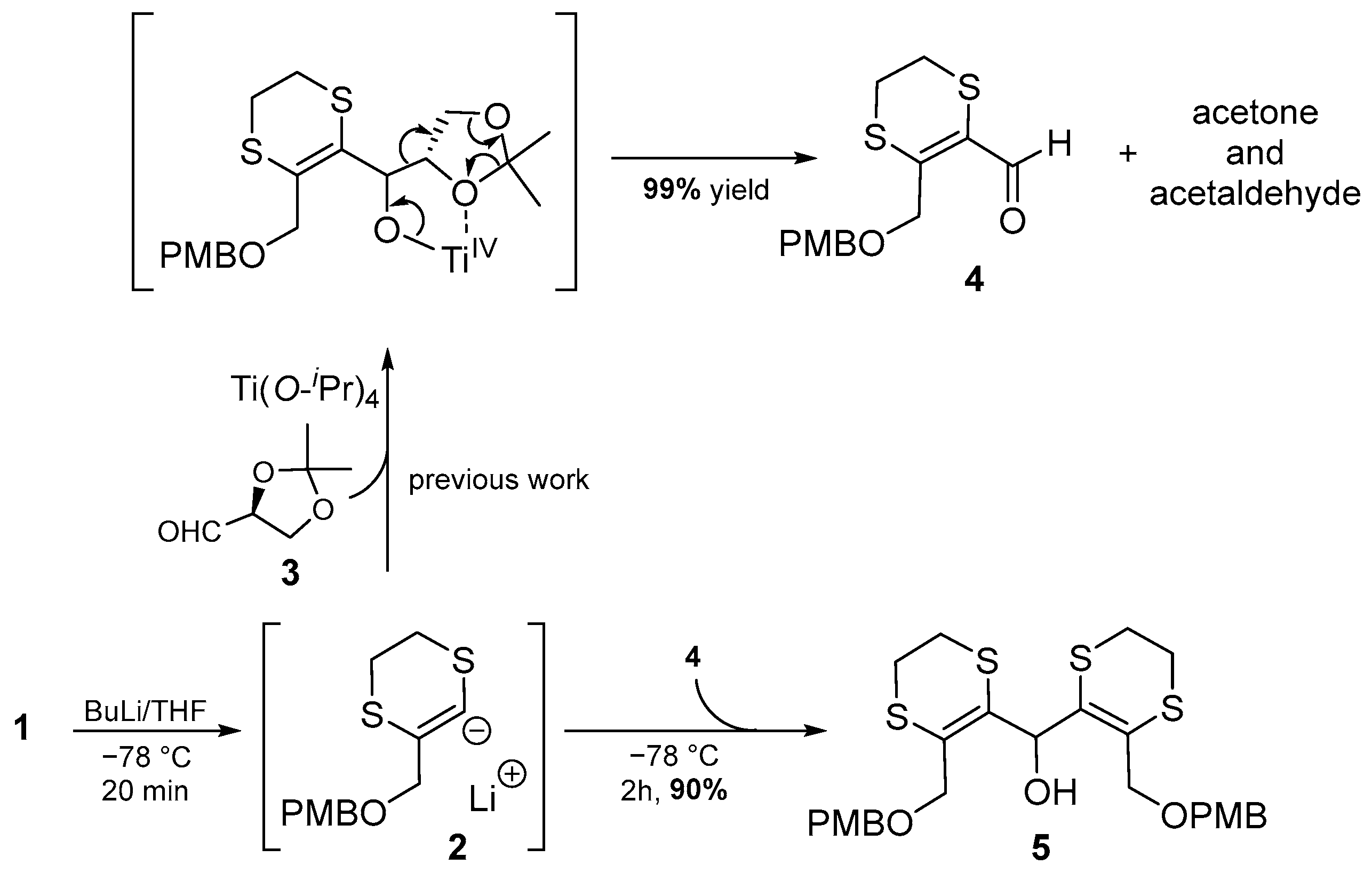
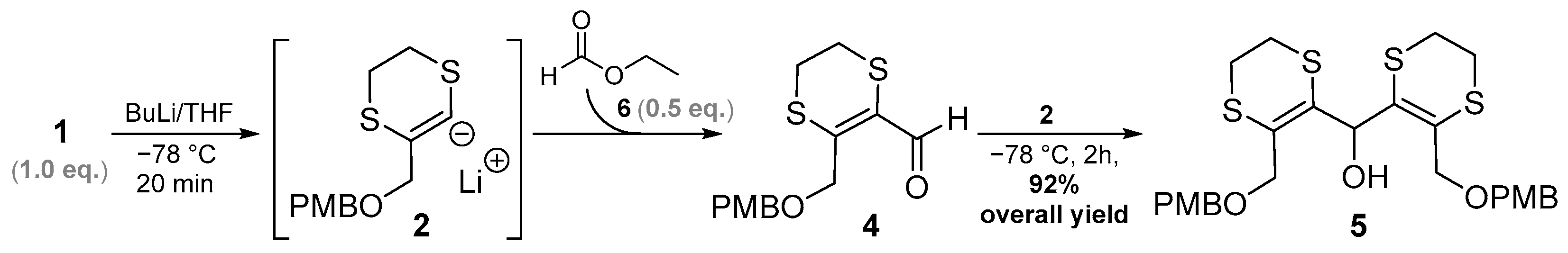
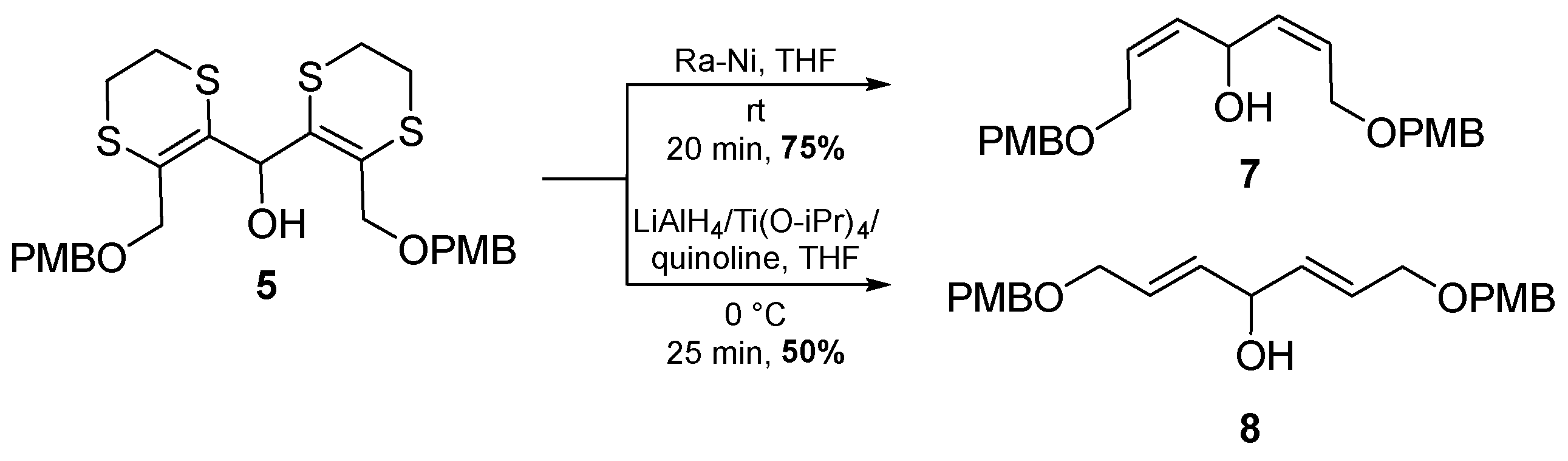
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Bis(3-(((4-methoxybenzyl)oxy)methyl)-5,6-dihydro-1,4-dithiin-2-yl)methanol (5)
3.2.1. Method A (Two-Step Procedure)
3.2.2. Method B (Tandem Procedure)
3.3. (2Z,5Z)-1,7-Bis[(4-methoxybenzyl)oxy]hepta-2,5-dien-4-ol (7)
3.4. (2E,5E)-1,7-Bis[(4-methoxybenzyl)oxy]hepta-2,5-dien-4-ol (8)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stowell, J.C. Three-Carbon Homologating Agents. Chem. Rev. 1984, 84, 409–435. [Google Scholar] [CrossRef]
- Pace, V. (Ed.) Homologation Reactions; Wiley-VCH (Verlag): New York, NY, USA, 2023; ISBN 9783527348152. [Google Scholar]
- Dondoni, A. Advances in the Use of Synthons in Organic Chemistry; Elsevier: Amsterdam, The Netherlands, 1993; ISBN 9781483100944. [Google Scholar]
- Candeias, N.R.; Paterna, R.; Gois, P.M.P. Homologation Reaction of Ketones with Diazo Compounds. Chem. Rev. 2016, 116, 2937–2981. [Google Scholar] [CrossRef]
- Li, J.J. (Ed.) Name Reactions for Homologations, Part I; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; ISBN 9780470487020. [Google Scholar]
- Ansari, A.; Kumar, V. (Eds.) Recent Advances in Organometallic Chemistry; Elsevier: Amsterdam, The Netherlands, 2024; ISBN 9780323905961. [Google Scholar] [CrossRef]
- Kotha, S.; Meshram, M. Application of Organometallics in Organic Synthesis. J. Organomet. Chem. 2018, 874, 13–25. [Google Scholar] [CrossRef]
- Castoldi, L.; Monticelli, S.; Senatore, R.; Ielo, L.; Pace, V. Homologation Chemistry with Nucleophilic α-Substituted Organometallic Reagents: Chemocontrol, New Concepts and (Solved) Challenges. Chem. Commun. 2018, 54, 6692–6704. [Google Scholar] [CrossRef] [PubMed]
- Afarinkia, K. Main Group Organometallics in Organic Synthesis. J. Chem. Soc. Perkin Trans. 1 1999, 15, 2025–2046. [Google Scholar] [CrossRef]
- Coldham, I. Main Group Organometallics in Synthesis. J. Chem. Soc. Perkin Trans. 1 1998, 4, 1343–1364. [Google Scholar] [CrossRef]
- Hartley, F.R.; Patai, S. (Eds.) The Metal-Carbon Bond; John Wiley & Sons, Inc.: Chichester, UK, 1987; Volume 4, ISBN 9780470771778. [Google Scholar]
- Frenking, G.; Fröhlich, N. The Nature of the Bonding in Transition-Metal Compounds. Chem. Rev. 2000, 100, 717–774. [Google Scholar] [CrossRef]
- Haiduc, I.; Silaghi-Dumitrescu, L. 2 Organometallic Molecules: The Nature of Metal–Carbon Bonds. In Organometallic Chemistry; Haiduc, I., Ed.; De Gruyter: Berlin, Germany, 2022; pp. 5–18. [Google Scholar]
- Najera, C.; Yus, M. Functionalized Organolithium Compounds: New Synthetic Adventures. Curr. Org. Chem. 2003, 7, 867–926. [Google Scholar] [CrossRef]
- Rappoport, Z.; Marek, I. (Eds.) The Chemistry of Organolithium Compounds; Wiley: Hoboken, NJ, USA, 2004; ISBN 9780470843390. [Google Scholar]
- Gawley, R.E. Overview of Carbanion Dynamics and Electrophilic Substitutions in Chiral Organolithium Compounds. In Stereochemical Aspects of Organolithium Compounds; John Wiley & Sons: Hoboken, NJ, USA, 2010; pp. 93–133. [Google Scholar]
- Power, M.; Alcock, E.; McGlacken, G.P. Organolithium Bases in Flow Chemistry: A Review. Org. Process Res. Dev. 2020, 24, 1814–1838. [Google Scholar] [CrossRef]
- Luisi, R.; Capriati, V. (Eds.) Lithium Compounds in Organic Synthesis; Wiley: Hoboken, NJ, USA, 2014; ISBN 9783527333431. [Google Scholar]
- Beak, P.; Basu, A.; Gallagher, D.J.; Park, Y.S.; Thayumanavan, S. Regioselective, Diastereoselective, and Enantioselective Lithiation−Substitution Sequences: Reaction Pathways and Synthetic Applications. Acc. Chem. Res. 1996, 29, 552–560. [Google Scholar] [CrossRef]
- Guijarro, D.; M Pastor, I.; Yus, M. Non-Deprotonating Methodologies for Organolithium Reagents Starting from Non-Halogenated Materials. Part 1: Carbon—Heteroatom Bond Cleavage. Curr. Org. Chem. 2011, 15, 375–400. [Google Scholar] [CrossRef]
- De Fenza, M.; Esposito, A.; D’Alonzo, D.; Guaragna, A. Synthesis of Piperidine Nucleosides as Conformationally Restricted Immucillin Mimics. Molecules 2021, 26, 1652. [Google Scholar] [CrossRef]
- Loreto, D.; Esposito, A.; Demitri, N.; Guaragna, A.; Merlino, A. Digging into Protein Metalation Differences Triggered by Fluorine Containing-Dirhodium Tetracarboxylate Analogues. Dalton Trans. 2022, 51, 7294–7304. [Google Scholar] [CrossRef]
- Loreto, D.; Esposito, A.; Demitri, N.; Guaragna, A.; Merlino, A. Reactivity of a Fluorine-Containing Dirhodium Tetracarboxylate Compound with Proteins. Dalton Trans. 2022, 51, 3695–3705. [Google Scholar] [CrossRef] [PubMed]
- Esposito, A.; di Giovanni, C.; De Fenza, M.; Talarico, G.; Chino, M.; Palumbo, G.; Guaragna, A.; D’Alonzo, D. A Stereoconvergent Tsuji–Trost Reaction in the Synthesis of Cyclohexenyl Nucleosides. Chem. A Eur. J. 2020, 26, 2597–2601. [Google Scholar] [CrossRef]
- De Pasquale, V.; Esposito, A.; Scerra, G.; Scarcella, M.; Ciampa, M.; Luongo, A.; D’Alonzo, D.; Guaragna, A.; D’Agostino, M.; Pavone, L.M. N-Substituted l-Iminosugars for the Treatment of Sanfilippo Type B Syndrome. J. Med. Chem. 2023, 66, 1790–1808. [Google Scholar] [CrossRef]
- Esposito, A.; D’alonzo, D.; D’errico, S.; De Gregorio, E.; Guaragna, A. Toward the Identification of Novel Antimicrobial Agents: One-Pot Synthesis of Lipophilic Conjugates of N-Alkyl d-and l-Iminosugars. Mar. Drugs 2020, 18, 572. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Shi, Y. A New Type of Ketone Catalyst for Asymmetric Epoxidation. J. Org. Chem. 1997, 62, 8622–8623. [Google Scholar] [CrossRef]
- Ryckaert, B.; Demeyere, E.; Degroote, F.; Janssens, H.; Winne, J.M. 1,4-Dithianes: Attractive C2-Building Blocks for the Synthesis of Complex Molecular Architectures. Beilstein J. Org. Chem. 2023, 19, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Garner, P.; Park, J.M. 1,1,-Dimethylethyl (S)- or (R)-4-Formyl-2,2-Dimethyl-3-Oxazolidinecarboxylate: A Useful Serinal Derivative. Org. Synth. 1992, 70, 18. [Google Scholar] [CrossRef]
- Hubschwerlen, C.; Specklin, J.-L.; Higelin, J. l-(S)-Glyceraldehyde Acetonide. Org. Synth. 1995, 72, 1. [Google Scholar] [CrossRef]
- Schmid, C.R.; Bryant, J.D. d-(R)-Glyceraldehyde Acetonide. Org. Synth. 1995, 72, 6. [Google Scholar] [CrossRef]
- Hodgson, R.; Majid, T.; Nelson, A. Towards Complete Stereochemical Control: Complementary Methods for the Synthesis of Six Diastereoisomeric Monosaccharide Mimetics. J. Chem. Soc. Perkin Trans. 1 2002, 12, 1444–1454. [Google Scholar] [CrossRef]
- Allart, B.; Busson, R.; Rozenski, J.; Van Aerschot, A.; Herdewijn, P. Synthesis of Protected d-Altritol Nucleosides as Building Blocks for Oligonucleotide Synthesis. Tetrahedron 1999, 55, 6527–6546. [Google Scholar] [CrossRef]
- Rentner, J.; Kljajic, M.; Offner, L.; Breinbauer, R. Recent Advances and Applications of Reductive Desulfurization in Organic Synthesis. Tetrahedron 2014, 70, 8983–9027. [Google Scholar] [CrossRef]
- Becker, S.; Fort, Y.; Caubere, P. New Desulfurizations by Nickel-Containing Complex Reducing Agents. J. Org. Chem. 1990, 55, 6194–6198. [Google Scholar] [CrossRef]
- Caputo, R.; Palumbo, G.; Pedatella, S. Diastereoselective Desulfurization of 5,6-Dihydro-1,4-Dithiins. Synthesis of Muscalure from Musca domestica L. Tetrahedron 1994, 50, 7265–7268. [Google Scholar] [CrossRef]
- Kramer, R.; Berkenbusch, T.; Brückner, R. Stereocomplementary Desymmetrizations of Divinylcarbinols by Zirconium(IV)- vs. Titanium(IV)-Mediated Asymmetric Epoxidations. Adv. Synth. Catal. 2008, 350, 1131–1148. [Google Scholar] [CrossRef]
- Nachbauer, L.; Brückner, R. Synthesis of a Cn–Cn+6 Building Block Common to Important Polyol,Polyene Antibiotics from a Divinylcarbinol by a Desymmetrizing Sharpless Epoxidation. Eur. J. Org. Chem. 2013, 2013, 6545–6562. [Google Scholar] [CrossRef]
- Trost, B.M.; Wrobleski, S.T.; Chisholm, J.D.; Harrington, P.E.; Jung, M. Total Synthesis of (+)-Amphidinolide A. Assembly of the Fragments. J. Am. Chem. Soc. 2005, 127, 13589–13597. [Google Scholar] [CrossRef]
- Kramer, R.; Brückner, R. Stereocontrolled Synthesis of a Cn–Cn+6 Building Block for the Unnatural Enantiomers of Important Polyol,Polyene Antibiotics from an Epoxy Alcohol by a Reduction/Conjugate Addition/Hydroxylation Sequence. Eur. J. Org. Chem. 2013, 2013, 6563–6583. [Google Scholar] [CrossRef]
- De Angelis, M.; Primitivo, L.; Sappino, C.; Centrella, B.; Lucarini, C.; Lanciotti, L.; Petti, A.; Odore, D.; D’Annibale, A.; Macchi, B.; et al. Stereocontrolled Synthesis of New Iminosugar Lipophilic Derivatives and Evaluation of Biological Activities. Carbohydr. Res. 2023, 534, 108984. [Google Scholar] [CrossRef] [PubMed]
- Somfai, P.; Marchand, P.; Torsell, S.; Lindström, U.M. Asymmetric Synthesis of (+)-1-Deoxynojirimycin and (+)-Castanospermine. Tetrahedron 2003, 59, 1293–1299. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esposito, A.; Guaragna, A. Bis(3-(((4-methoxybenzyl)oxy)methyl)-5,6-dihydro-1,4-dithiin-2-yl)methanol. Molbank 2024, 2024, M1867. https://doi.org/10.3390/M1867
Esposito A, Guaragna A. Bis(3-(((4-methoxybenzyl)oxy)methyl)-5,6-dihydro-1,4-dithiin-2-yl)methanol. Molbank. 2024; 2024(3):M1867. https://doi.org/10.3390/M1867
Chicago/Turabian StyleEsposito, Anna, and Annalisa Guaragna. 2024. "Bis(3-(((4-methoxybenzyl)oxy)methyl)-5,6-dihydro-1,4-dithiin-2-yl)methanol" Molbank 2024, no. 3: M1867. https://doi.org/10.3390/M1867
APA StyleEsposito, A., & Guaragna, A. (2024). Bis(3-(((4-methoxybenzyl)oxy)methyl)-5,6-dihydro-1,4-dithiin-2-yl)methanol. Molbank, 2024(3), M1867. https://doi.org/10.3390/M1867