
(S)-1-Methyl-2-oxoimidazolidine-4-carboxylic Acid


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
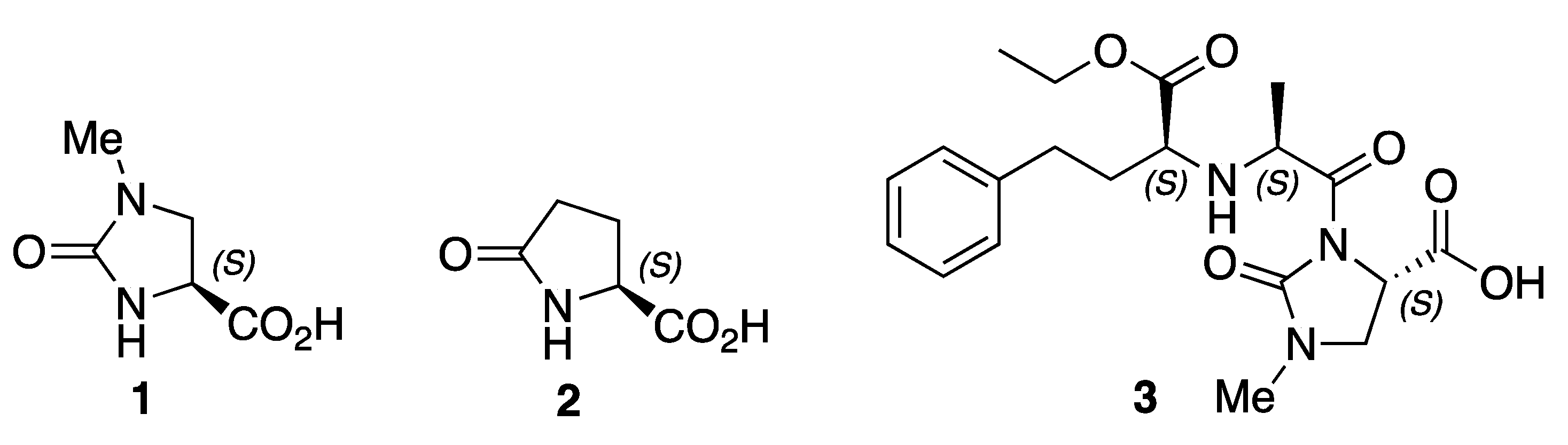
1. Introduction
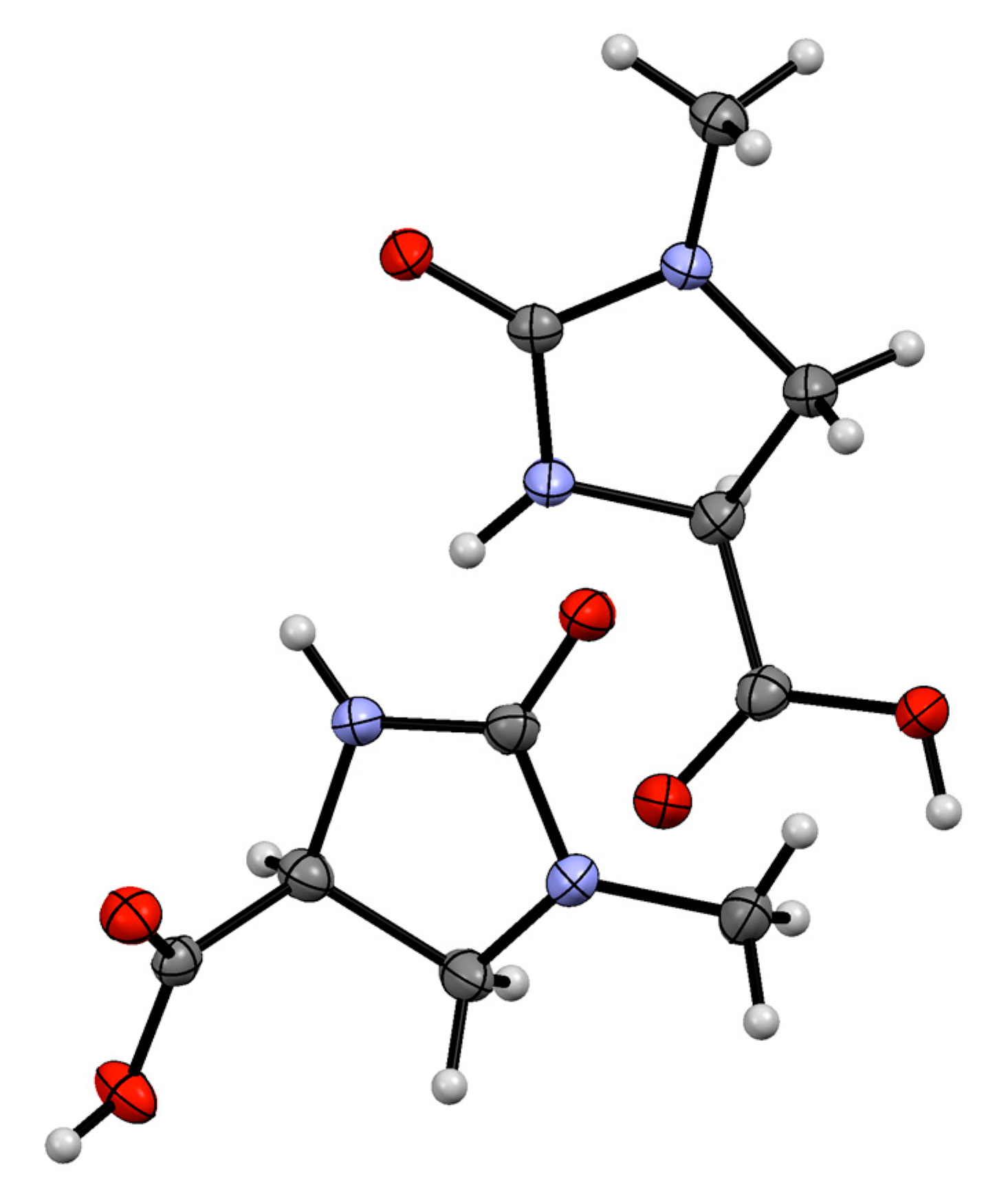
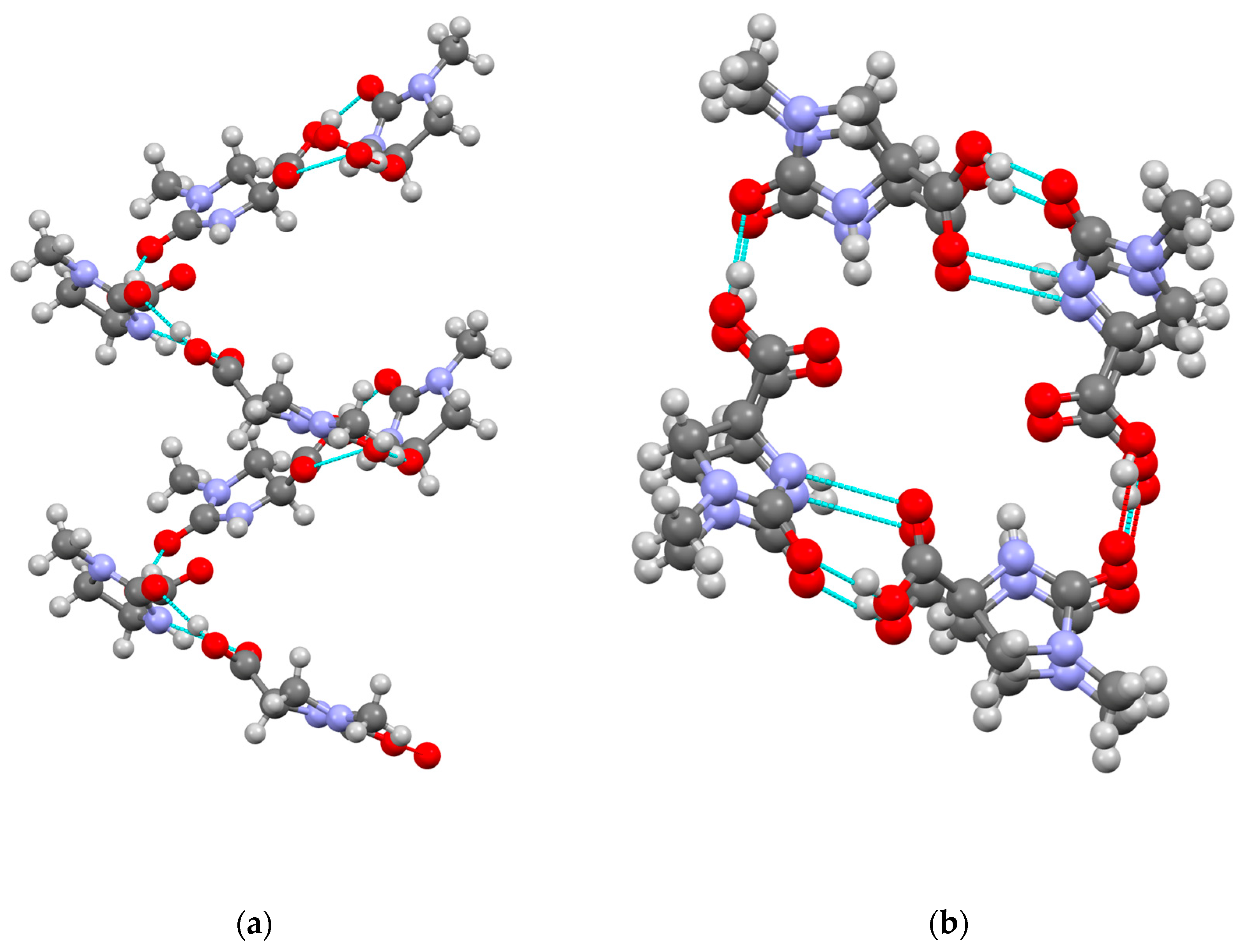
2. Results
3. Experimental
3.1. General Experimental Details
3.2. Synthesis of (S)-1-Methyl-2-oxoimidazolidine-4-carboxylic Acid 1
3.3. X-ray Structure Determination of 1
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bøler, J.; Enzmann, F.; Folkers, K.; Bowers, C.Y.; Schally, A.V. The identity of chemical and hormonal properties of the thyrotropin releasing hormone and pyroglutamyl-histidyl-proline amide. Biochem. Biophys. Res. Commun. 1969, 37, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Seeburg, P.H.; Adelman, J.P. Characterization of cDNA for precursor of human luteinizing hormone releasing hormone. Nature 1984, 311, 666–668. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Nunami, K.; Kato, J.; Yoneda, N.; Kubo, M.; Ochiai, T.; Ishida, R. Studies of Angiotensin Converting Enzyme Inhibitors. 4. Synthesis and Angiotensin Converting Inhibitory Activities of 3-Acyl-1-alkyl-2-oxoimidazolidine-4-carboxylic Acid Derivatives. J. Med. Chem. 1989, 32, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, M.; Horimoto, S.; Mabuchi, M.; Banno, K. Determination of three metabolites of a new angiotensin-converting enzyme inhibitor, imidapril, in plasma and urine by gas chromatography-mass spectrometry using multiple ion detection. J. Chromatograph. 1992, 581, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.M.; Curran, M.P.; Lyseng-Williamson, K.A. Imidapril A Review of its Use in Essential Hypertension, Type 1 Diabetic Nephropathy and Chronic Heart Failure. Drugs 2007, 67, 1359–1378. [Google Scholar] [CrossRef] [PubMed]
- Beswick, P.J.; Dean, D.K.; Gleave, R.J.; Moses, A.P. Walter DS. Imidazolidine Carboxamide Derivatives as P2X7 Modulators. World Patent WO2008119825A2, 9 October 2008. [Google Scholar]
- Lemieux, R.M.; Popovici-Muller, J.; Travins, J.M.; Cai, Z.; Cui, D.; Zhou, D. Therapeutically Active Compounds and Their Methods of Use. World Patent WO2015010297A1, 29 January 2015. [Google Scholar]
- Gai, Y.; Niu, D.; Or, Y.S.; Wang, Z. Cyclic P3 Tripeptide Hepatitis C Serine Protease Inhibitors. US Patent 2008/0267916 A1, 30 October 2008. [Google Scholar]
- Saijo, S.; Wada, M.; Himizu, J.-I.; Ishida, A. Heterocyclic Prostaglandins. VI. Synthesis of 11-Deoxy-8,10-diazaprostaglandin E1 and its 10-Methyl Derivative. Chem. Pharm. Bull. 1980, 28, 1459–1467. [Google Scholar] [CrossRef]
- Panda, M.K.; Runčevski Husain, A.; Dinnebier, R.E.; Naumov, P. Perpetually Self-Propelling Chiral Single Crystals. J. Am. Chem. Soc. 2015, 137, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
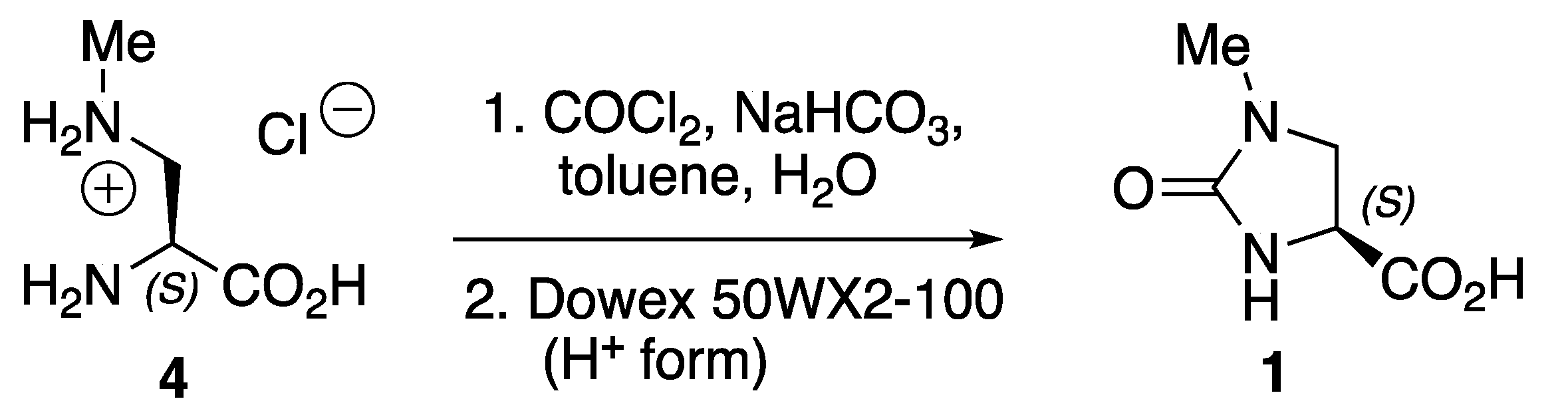
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dey, A.L.; Motevalli, M.; Abrahams, I.; Wyatt, P.B. (S)-1-Methyl-2-oxoimidazolidine-4-carboxylic Acid. Molbank 2024, 2024, M1835. https://doi.org/10.3390/M1835
Dey AL, Motevalli M, Abrahams I, Wyatt PB. (S)-1-Methyl-2-oxoimidazolidine-4-carboxylic Acid. Molbank. 2024; 2024(2):M1835. https://doi.org/10.3390/M1835
Chicago/Turabian StyleDey, Ashley L., Majid Motevalli, Isaac Abrahams, and Peter B. Wyatt. 2024. "(S)-1-Methyl-2-oxoimidazolidine-4-carboxylic Acid" Molbank 2024, no. 2: M1835. https://doi.org/10.3390/M1835
APA StyleDey, A. L., Motevalli, M., Abrahams, I., & Wyatt, P. B. (2024). (S)-1-Methyl-2-oxoimidazolidine-4-carboxylic Acid. Molbank, 2024(2), M1835. https://doi.org/10.3390/M1835