1. Introduction
Porphyrins are a class of macrocyclic compounds with a wide range of applications. They are characterized by a planar structure described for the first time in 1912 [
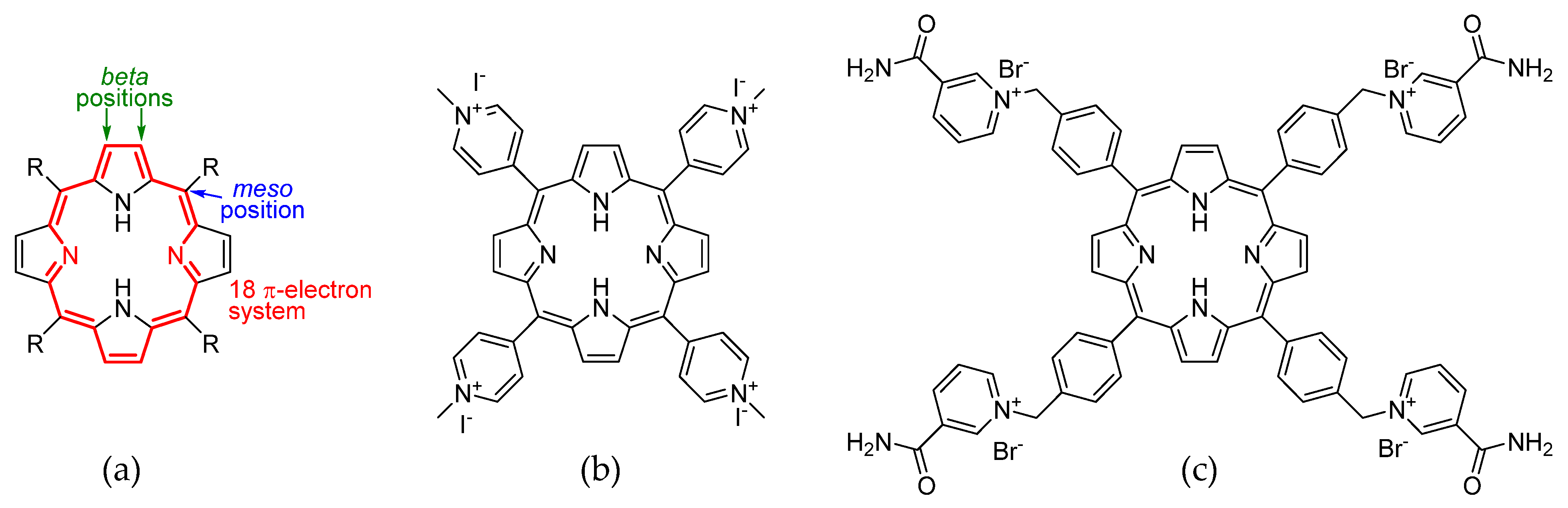
1]. This system contains 26 π-electrons, 18 of which contribute to the aromaticity. On the porphyrin core, two main positions for functionalization can be distinguished: the beta position on the pyrrole rings, and the methylene bridge, called the meso position (
Figure 1a). Porphyrins are obtained by a condensation between an aldehyde and pyrrole. The choice of a suitable aldehyde provides a meso substituent that can be used to modulate various chemical–physical, electronic, and spectroscopic properties of the macrocycle. Generally, porphyrins are soluble in organic solvents and practically insoluble in water. But, by placing specific meso substituents, it is possible to make them tetracationic. To the contrary of the neutral form, the salt form shows more hydrophilicity, making porphyrins useful in aqueous solution applications. One of the best known tetracationic porphyrins in the literature is a derivative of the 5,10,15,20-tetra(4-pyridyl)porphyrin (TPyP) 5,10,15,20-tetra(
N-methylpyridinium-4-yl)porphyrin (TMPyP4) (
Figure 1b), which was proposed in 1998 [
2] as a stabilizer for non-canonical quaternary DNA structures, the G-quadruplexes (G4). The potential interest in G4 in the field of cancer therapy is suggested by their formation in the presence of K
+ or Na
+ ions from guanine-rich sequences, which include those found in the telomeric region that makes them suitable as telomerase inhibitors, or in protooncogene domains, including NHEIII
1 within c-Myc [
3,
4,
5]. The structure of TMPyP4 involves the use of the meso-position pyridine nitrogen on the ring to form the ammonium group via a nucleophilic substitution reaction with iodomethane [
6]. In this way, the cationic portions are close to the ligand core. In general, porphyrin ligands tend to interact with G4 by stacking on G-quartets [
7]; therefore, the distancing of the cationic moiety from the core was set up so that it could fold and interact with the loops of G4. Here we propose the synthesis of 5,10,15,20-tetrakis-(4-(3-carbamoyl-pyridyl)-methylphenyl)porphyrin bromide (P15p), a new tetracationic porphyrin derivative that shows high solubility in water and thus a possible application as a ligand of G4 (
Figure 1c).
2. Results and Discussion
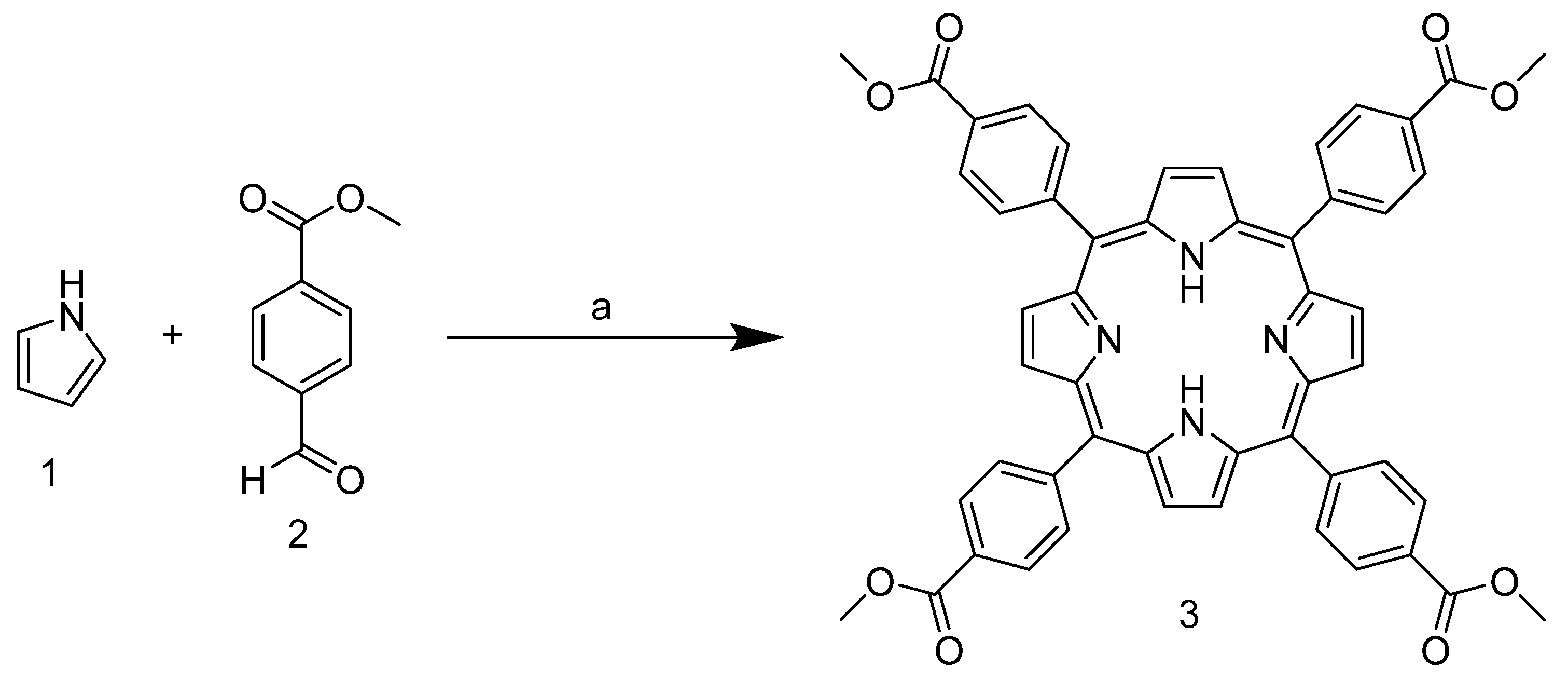
As mentioned above, the tetracationic porphyrin derivatives currently best known in the literature and studied as stabilizers are derived from TPyP. In our case, however, the starting point is 5,10,15,20-tetrakis-(4-carbomethoxyphenyl)porphyrin
3, which has benzoate substituents in the meso position. This can be obtained by condensation reaction between pyrrole and 4-formylbenzoate in xylene at reflux temperature (
Scheme 1).
Due to the presence of the ester group, which subtracts the electronic density and stabilizes the carbocation in the
para position, the aldehyde carbonyl of
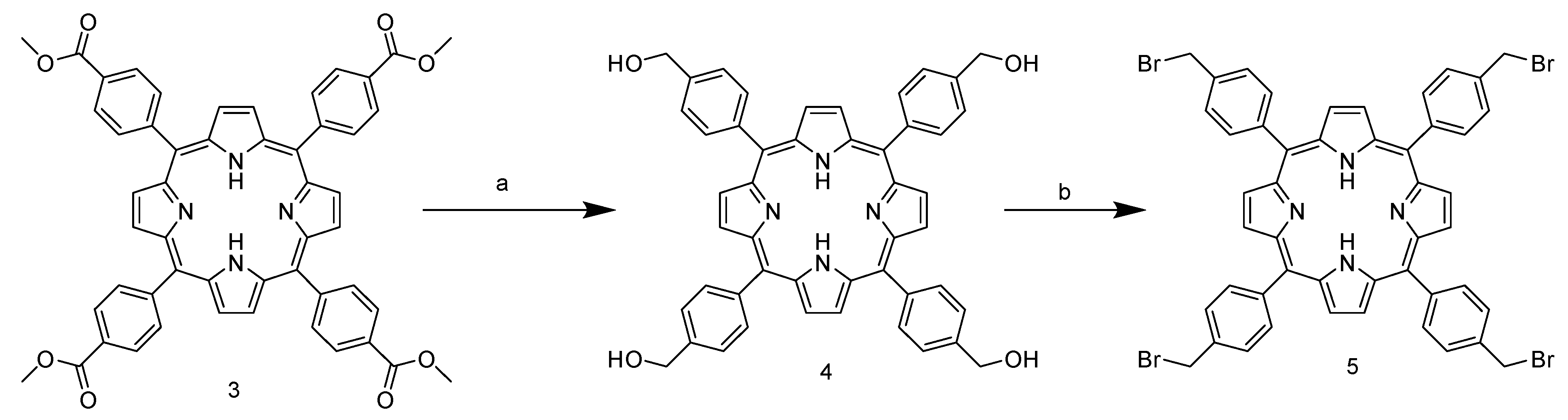
2 shows high electrophilicity, yielding nearly 40% of the product, and is above the average yield in porphyrin synthesis. In addition, the use of a non-halogenated solvent and the choice of an inexpensive and readily available catalyst enhance the greenness of the process. The next step involves the ester group reduction using LiAlH
4 in THF. The resulting alcohol group is then used as a starting point for the halogenation reaction with PBr
3 (
Scheme 2).
Unlike the previous steps, the tetra-halogenated product
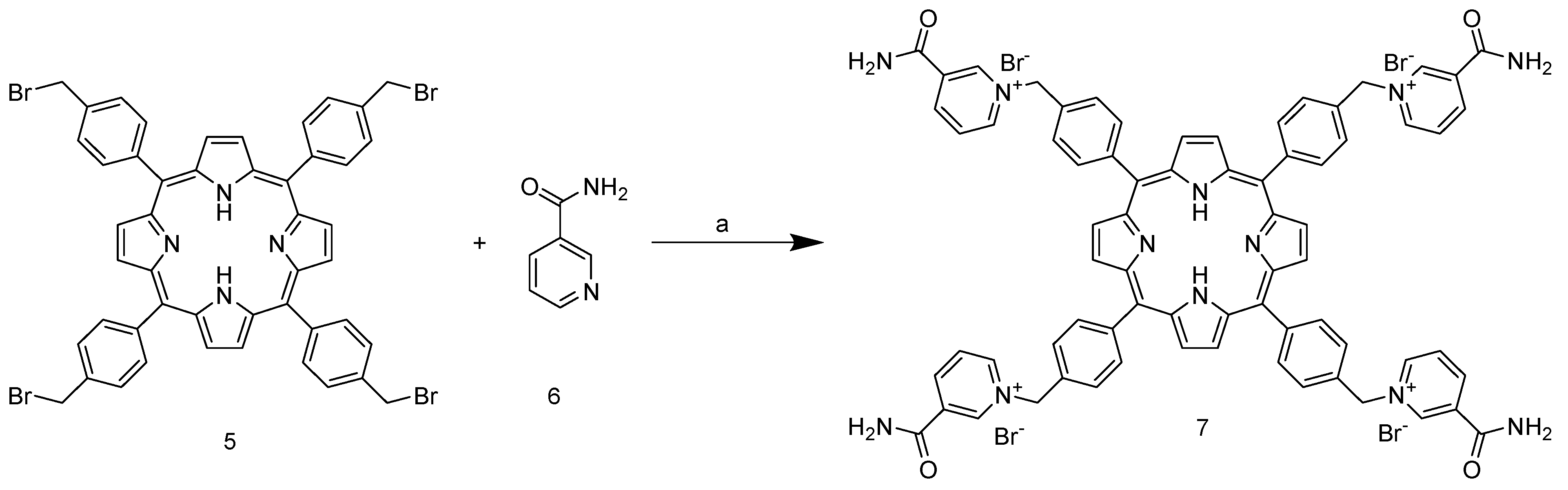
5 requires chromatographic purification on silica with DCM as the eluent. The final step involves nucleophilic substitution on the pyridine nitrogen of nicotinamide (
Scheme 3).
The reaction is carried out in DMF at 150 °C and leads to the isolable tetracationic product by precipitation with DCM. The use of a large excess of nicotinamide prevents partial substitution; moreover, our experiments show that once the co-solvent for precipitation is added, only the tetrasubstituted product tends to precipitate, as shown in the NMR spectra (
Figure S4). Cleaning product with acetone removes all residues of nicotinamide from the filter and also offers the possibility to recover it. The end of the reaction can be assessed via a partitioning experiment by adding a drop of reaction solution to H
2O/DCM 1:1
v/
v mixture. The colored aqueous phase indicates product formation (
Figure 2).
3. Materials and Methods
All reagents and chemicals were sourced from commercial suppliers and utilized without additional purification: lithium aluminium hydride (Acros Organics, 95%, Geel, Belgium), Nicotinamide (Sigma Aldrich, >98%, Merck KGaA, Darmstadt, Germany), Phosphorus Tribromide (Sigma Aldrich, >97%), Pyrrole (TCI Chemicals, >99%, Tokyo, Japan), N,N-dimethylformamide (Sigma Aldrich, 99.8%), and Tetrahydrofuran (VWR, 99%, Radnor, PA, USA). NMR spectral data were collected on a Bruker Avance Neo 400 spectrometer (Bruker, Ettlingen, Germany) operating at 400 MHz for 1H NMR and 101 MHz for 13C NMR. Measurements were conducted at room temperature (approximately 295 K). MS analysis was performed on a Thermo Finnigan Q-Exactive mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) equipped with an API-HESI source and Fourier Transform.
3.1. Synthesis of 5,10,15,20-Tetrakis-(4-carbomethoxyphenyl)porphyrin 3
In an inert atmosphere, 2.2 g of salicylic acid (16 mmol) and 5.2 g of methyl-4-formyl benzoate (32 mmol, 2 eq) were dissolved in 130 mL of xylene. The reaction mixture was heated to reflux, and 2.2 g of pyrrole (32 mmol, 1 eq) was added dropwise. After the addition, the mixture was refluxed for 2.5 h and then cooled to room temperature. Next, 15 mL of methanol was added, and the mixture was left to stand for 12 h. The black precipitate was filtrated and washed with methanol until the wash was almost colorless (purple powder, 2.42 g, 36%).
1H NMR (400 MHz, Chloroform-d) δ 8.82 (s, 8H), 8.45 (d, J = 6.6 Hz, 8H), 8.30 (d, J = 6.7 Hz, 8H), 4.12 (s, 12H), −2.80 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 167.38, 146.78, 134.67, 128.14, 52.61.
3.2. Synthesis of 5,10,15,20-Tetrakis-(4-hydroxymethylphenyl)porphyrin 4
To a suspension containing 1.1 g of LiAlH4 (28.5 mmol, 35 eq) in 63 mL of anhydrous THF, a solution of 0.5 g meso-tetra(4-carbomethoxyphenyl) porphyrin (0.81 mmol) in 212 mL anhydrous THF was added slowly under constant stirring. The system was then brought to reflux and after 1 h an additional 150 mL of anhydrous THF was added and the reaction was refluxed for a further 30 min. After cooling in an ice bath, the reaction mixture was quenched with water saturated Et2O to neutralize the excess LIAlH4. The mixture was filtered through celite, and the resulting solid was washed with THF until a nearly colorless filtrate was obtained. The porphyrin was extracted from the celite using a Soxhlet apparatus with a mixture of MeOH and THF in a 1:1 ratio. The solvents were then removed obtaining product (purple powder, 217 mg, 36%).
1H NMR (400 MHz, DMSO-d6) δ 8.84 (s, 8H), 8.17 (d, J = 5.6 Hz, 8H), 7.77 (d, J = 6.5 Hz, 8H), 5.55–5.44 (m, 4H), 4.88 (d, J = 5.6 Hz, 8H), −2.91 (s, 2H).
3.3. Synthesis of 5,10,15,20-Tetrakis-(4-bromomethylphenyl)porphyrin 5
An amount of 0.200 g of meso-tetra(4-hydroxymethylphenyl) porphyrin (0.27 mmol) was dissolved in 5 mL of anhydrous THF. Additionally, 90.5 µL of PBr
3 (0.95 mmol, 3.5 eq) was added dropwise to the solution while keeping the flask in an ice bath under an inert argon atmosphere. The reaction was allowed to proceed for 18 h at room temperature. After this time, the reaction mixture was quenched with 30 mL of saturated aqueous NaHCO
3 solution and stirred for 10 min. The product was extracted from the aqueous phase using 4 × 30 mL DCM. The resulting organic phases were dried with Na
2SO
4 and the solvents removed using Rotavapor. The reaction product was purified by chromatographic column with DCM as eluent (purple powder, 30 mg, 11%) [
8].
1H NMR (400 MHz, Chloroform-d) δ 8.84 (s, 8H), 8.19 (d, J = 8.1 Hz, 8H), 7.79 (d, J = 8.2 Hz, 8H), 4.86 (s, 8H), −2.81 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 142.36, 137.54, 135.07, 127.62, 119.69, 33.62.
3.4. Synthesis of 5,10,15,20-Tetrakis-(4-(3-carbamoyl-pyridyl)-methylphenyl)porphyrin bromide 7
The amount of 0.149 g of nicotinamide (1.22 mmol, 40 eq) was dissolved in 2 mL of DMF. Then, 30 mg of meso-tetra(4-bromomethylphenyl) porphyrin (0.03 mmol) was added. The reaction flask was capped and placed in an oil bath at 150 °C for 1 h. After cooling, 10 mL of DCM was added, resulting in the formation of a reddish-purple precipitate. The solid was collected by vacuum filtration and washed with acetone (purple powder, 63 mg, 90%).
1H NMR (400 MHz, DMSO-d6) δ 9.94 (s, 4H), 9.61 (d, J = 6.2 Hz, 4H), 9.12 (d, J = 8.2 Hz, 4H), 8.80 (s, 8H), 8.74 (s, 4H), 8.47 (dd, J = 8.2, 6.0 Hz, 4H), 8.33–8.26 (m, 12H), 8.06–7.97 (m, 8H), 6.31 (s, 8H), −2.99 (s, 2H). 13C NMR (101 MHz, DMSO) δ 162.83, 146.77, 145.35, 143.92, 142.06, 134.88, 134.34, 133.79, 128.51, 127.56, 119.35, 63.51. HRMS Electrospray ionization (ESI) m/z calcd for C72H58N12O44+ = 288.61705, found 288.61698. IR(ATR): 3674, 3036, 2897, 1703, 1401, 1183, 961, 797, 736, 677 cm−1. M.p.: 210 °C, decomposes.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}