4-(2-(5-(2-(tert-Butoxycarbonyl)hydrazinecarbonyl)-2-methylthiophen-3-yl)cyclopent-1-enyl)-5-methylthiophene-2-carboxylic Acid

Abstract
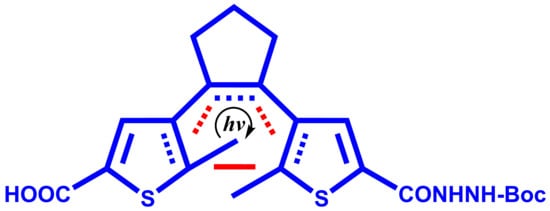
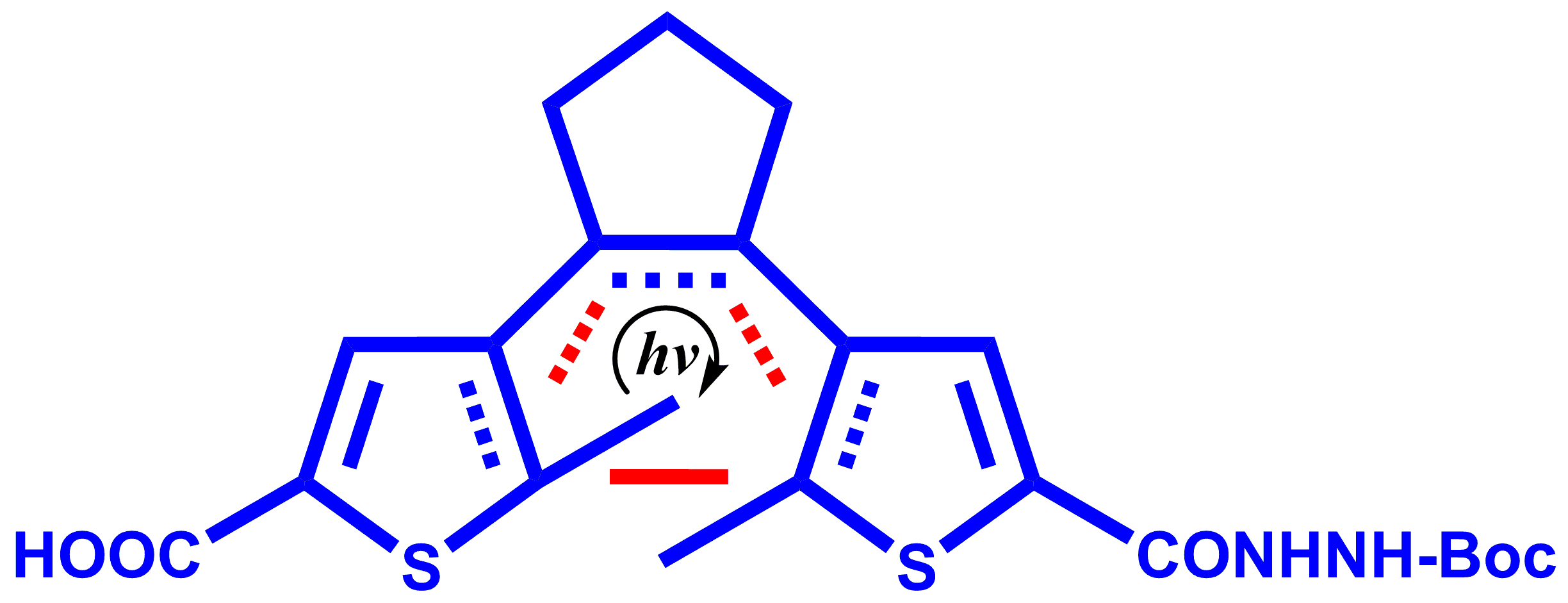
1. Introduction
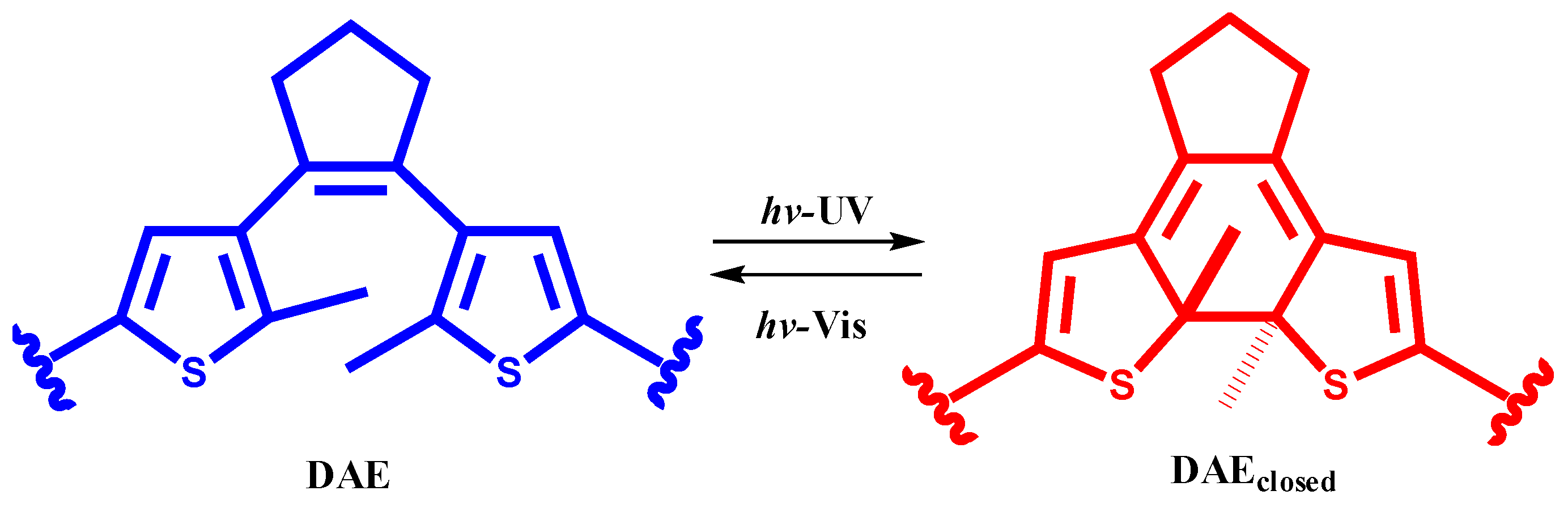

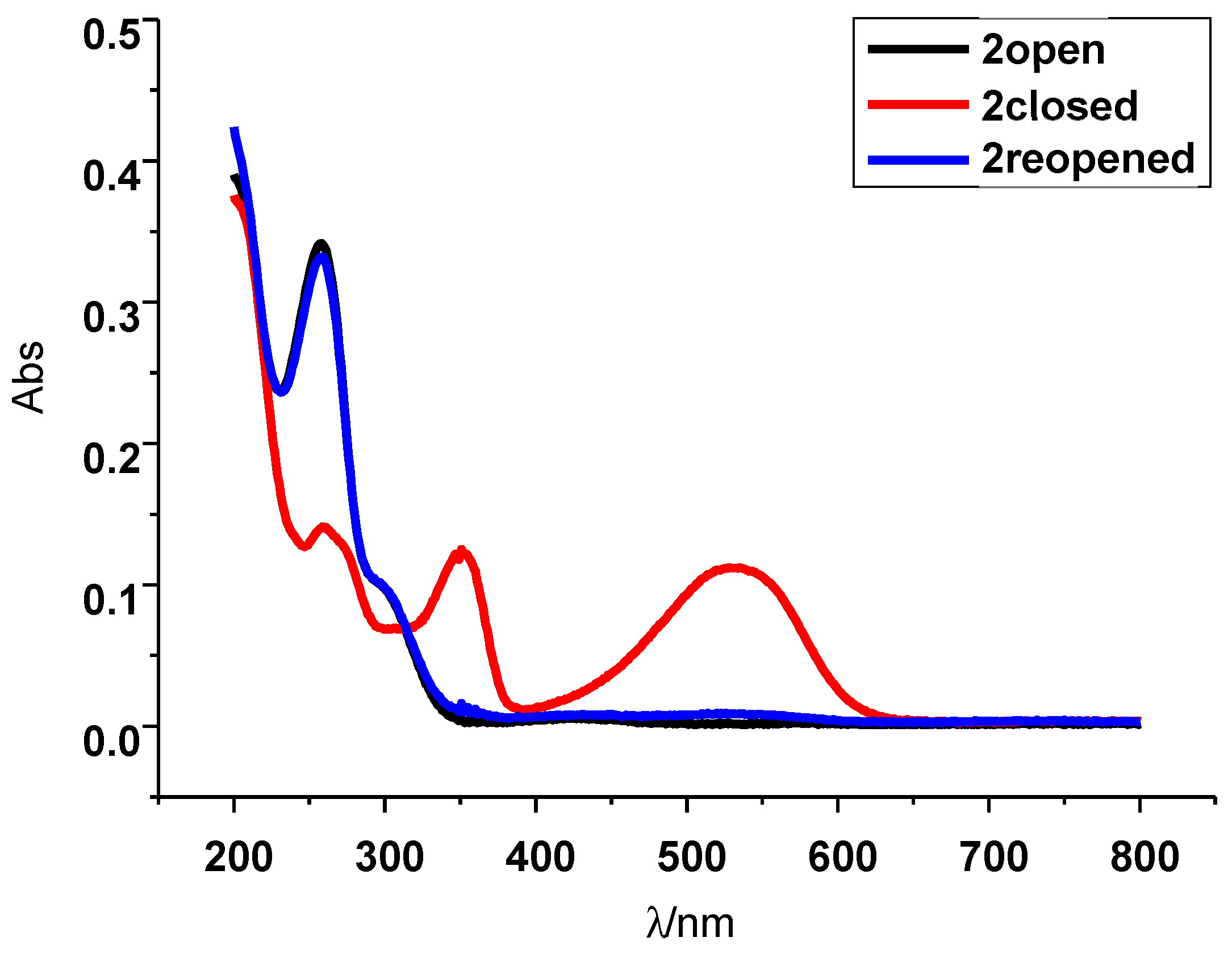
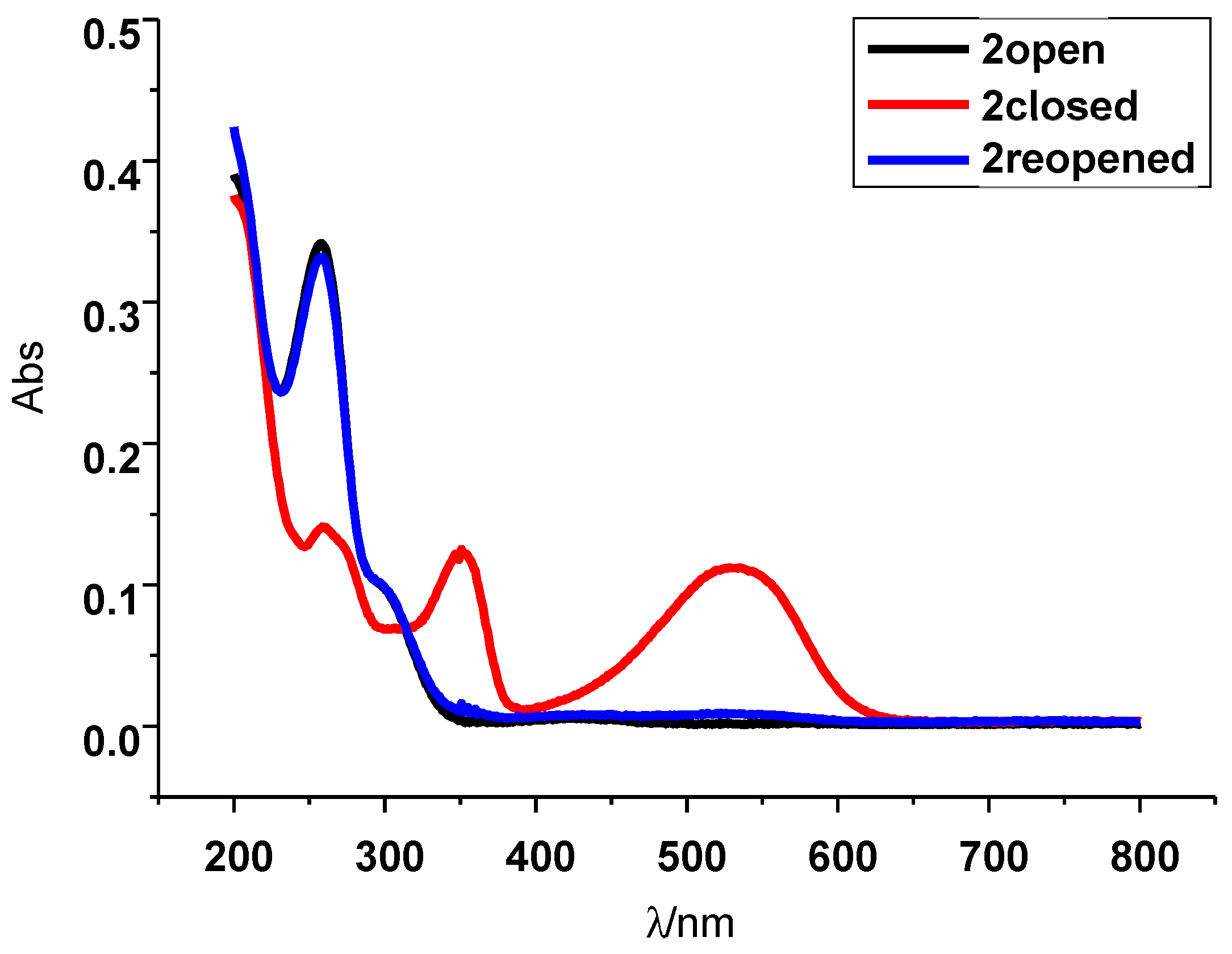
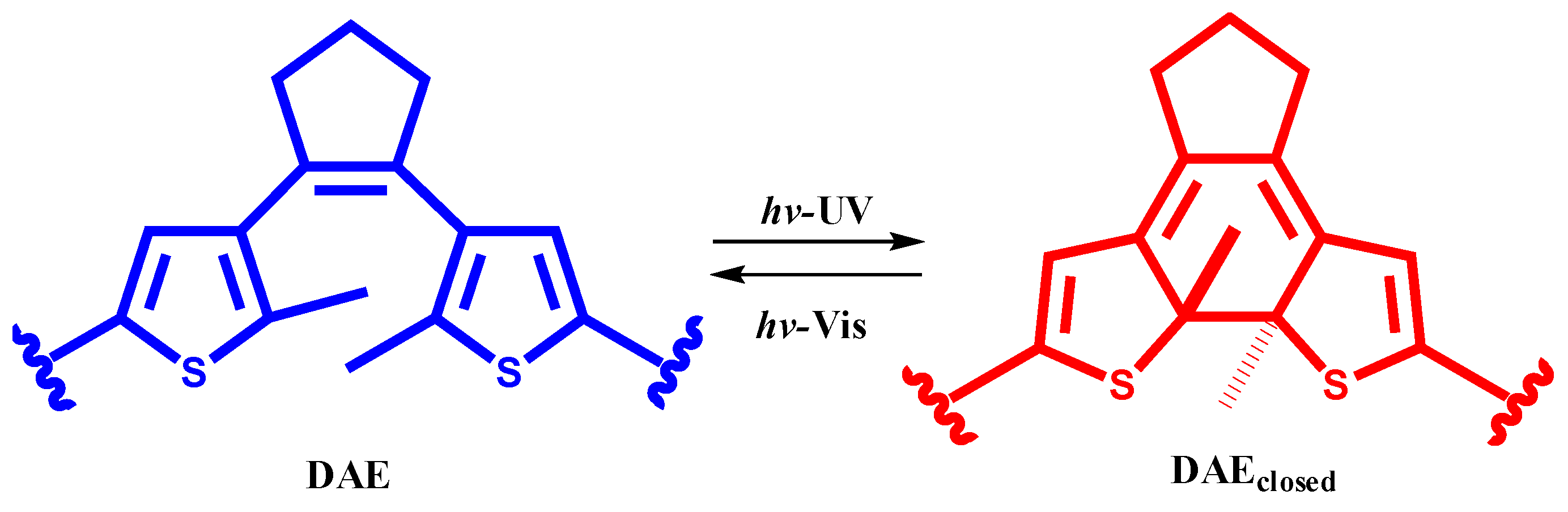
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Experimental
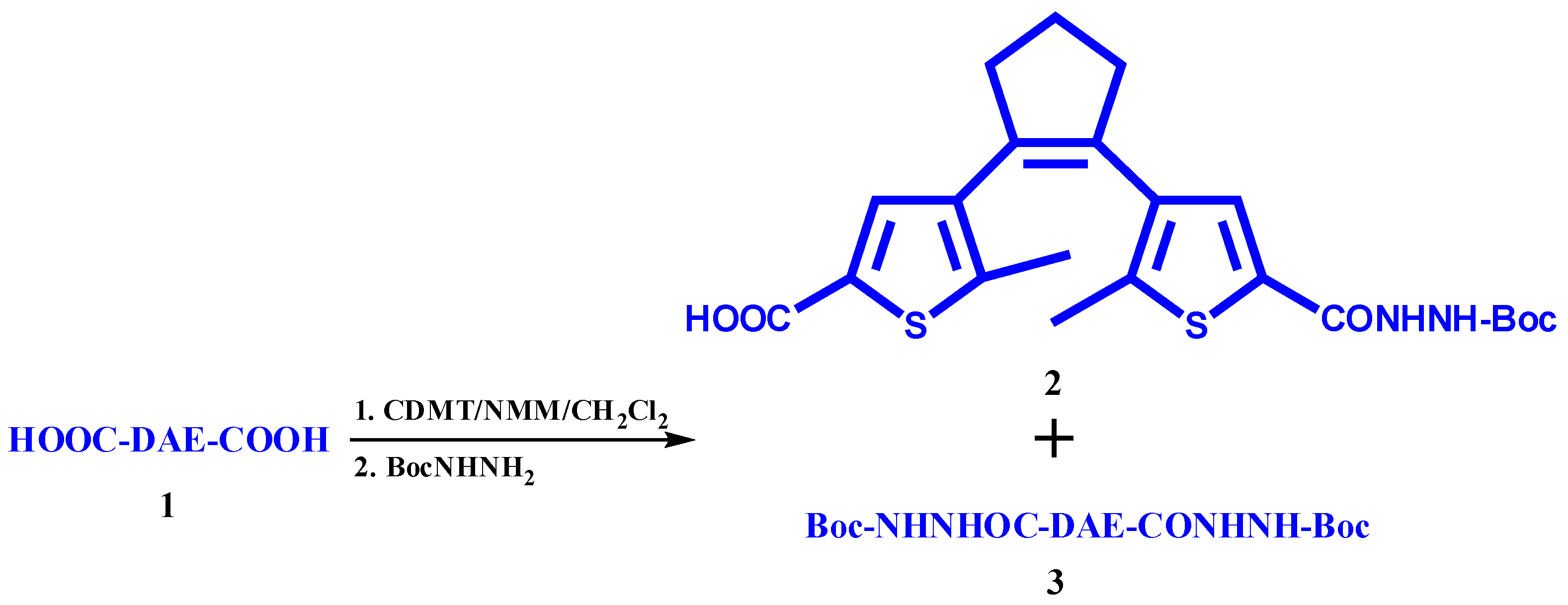
3.2.1. Synthesis of 4-(2-(5-(2-(tert-Butoxycarbonyl)hydrazinecarbonyl)-2-methylthiophen-3-yl)cyclopent-1-enyl)-5-methylthiophene-2-carboxylic Acid (2)
3.2.2. Details on by-Product 3 (Scheme 2)
3.2.3. Details on Closed Forms 2c and 3c
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Synthesis of 4-(2-(5-(2-(tert-butoxycarbonyl)hydrazinecarbonyl)-2-methylthiophen-3-yl)cyclopent-1-enyl)-2-(2-(tert-butoxycarbonyl)hydrazinecarbonyl)-5-methylthiophene (3)
Appendix A.2. Synthesis of 2c and 3c
Appendix B
References
- Albert, L.; Vázquez, O. Photoswitchable peptides for spatiotemporal control of biological functions. Chem. Commun. 2019, 55, 10192–10213. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, B.; Ahmed, S. Peptide-Based Low Molecular Weight Photosensitive Supramolecular Gelators. Gels. 2022, 8, 533. [Google Scholar] [CrossRef] [PubMed]
- Russew, M.-M.; Hecht, S. Photoswitches: From Molecules to Materials. Adv. Mater. 2010, 22, 3348–3360. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Irie, M. Diarylethene as a photoswitching unit. J. Photochem. Photobiol. C—Photochem. Rev. 2004, 5, 169–182. [Google Scholar] [CrossRef]
- Irie, M.; Fukaminato, T.; Matsuda, K.; Kobatake, S. Photochromism of Diarylethene Molecules and Crystals: Memories, Switches, and Actuators. Chem. Rev. 2014, 114, 12174–12277. [Google Scholar] [CrossRef] [PubMed]
- Irie, M. Diarylethene Molecular Photoswitches: Concepts and Functionalities, 1st ed.; Wiley-VCH: Weinheim, Germany, 2021. [Google Scholar]
- Cheng, H.-B.; Zhang, S.; Bai, E.; Cao, X.; Wang, J.; Qi, J.; Liu, J.; Zhao, J.; Zhang, L.; Yoon, J. Future-Oriented Advanced Diarylethene Photoswitches: From Molecular Design to Spontaneous Assembly Systems. Adv. Mater. 2022, 34, 2108289. [Google Scholar] [CrossRef] [PubMed]
- Babii, O.; Afonin, S.; Berditsch, M.; Reiber, S.; Mykhailiuk, P.K.; Kubyshkin, V.S.; Steinbrecher, T.; Ulrich, A.S.; Komarov, I.V. Controlling Biological Activity with Light: Diarylethene-Containing Cyclic Peptidomimetics. Angew. Chem. Int. Ed. 2014, 53, 3392–3395. [Google Scholar] [CrossRef] [PubMed]
- Orehovec, I.; Matković, M.; Pehar, I.; Majhen, D.; Piantanida, I. Bis-Pyrene Photo-Switch Open- and Closed-Form Differently Bind to ds-DNA, ds-RNA and Serum Albumin and Reveal Light-Induced Bioactivity. Int. J. Mol. Sci. 2021, 22, 4916. [Google Scholar] [CrossRef] [PubMed]
- Afonin, S.; Babii, O.; Komarov, I.; Mykhailiuk, P.; Ulrich, A. Preparation of Diarylethene-Containing Peptidomimetics Possessing Photo-Controlled Biological Activity; WO2014127919 A1; World Intellectual Property Organization: Geneva, Switzerland, 2014. [Google Scholar]
- van Dijken, D.J.; Beierle, J.M.; Stuart, M.C.A.; Szymański, W.; Browne, W.R.; Feringa, B.L. Autoamplification of Molecular Chirality through the Induction of Supramolecular Chirality. Angew. Chem. Int. Ed. 2014, 53, 5073–5077. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.ambeed.com/products/331432-79-2.html (accessed on 21 November 2023).
- Available online: https://www.ambeed.com/products/219537-97-0.html (accessed on 21 November 2023).
- Yamamura, S.; Toda, M.; Hirata, Y. Modified Clemmensen reduction: Cholestane. Org. Synth. 1973, 53, 86–89. [Google Scholar] [CrossRef]
- Available online: https://www.ambeed.com/products/219537-95-8.html (accessed on 27 November 2023).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagents | Isol./mg | Yield/% | Method of Isolation | |
|---|---|---|---|---|
| Fmoc | DIC/DIPEA/DMF | - | 50 1 | HPLC |
| Boc | CDMT/NMM/CH2Cl2 | 630 | 36 | Extraction/TLC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matković, M. 4-(2-(5-(2-(tert-Butoxycarbonyl)hydrazinecarbonyl)-2-methylthiophen-3-yl)cyclopent-1-enyl)-5-methylthiophene-2-carboxylic Acid. Molbank 2024, 2024, M1760. https://doi.org/10.3390/M1760
Matković M. 4-(2-(5-(2-(tert-Butoxycarbonyl)hydrazinecarbonyl)-2-methylthiophen-3-yl)cyclopent-1-enyl)-5-methylthiophene-2-carboxylic Acid. Molbank. 2024; 2024(1):M1760. https://doi.org/10.3390/M1760
Chicago/Turabian StyleMatković, Marija. 2024. "4-(2-(5-(2-(tert-Butoxycarbonyl)hydrazinecarbonyl)-2-methylthiophen-3-yl)cyclopent-1-enyl)-5-methylthiophene-2-carboxylic Acid" Molbank 2024, no. 1: M1760. https://doi.org/10.3390/M1760
APA StyleMatković, M. (2024). 4-(2-(5-(2-(tert-Butoxycarbonyl)hydrazinecarbonyl)-2-methylthiophen-3-yl)cyclopent-1-enyl)-5-methylthiophene-2-carboxylic Acid. Molbank, 2024(1), M1760. https://doi.org/10.3390/M1760