
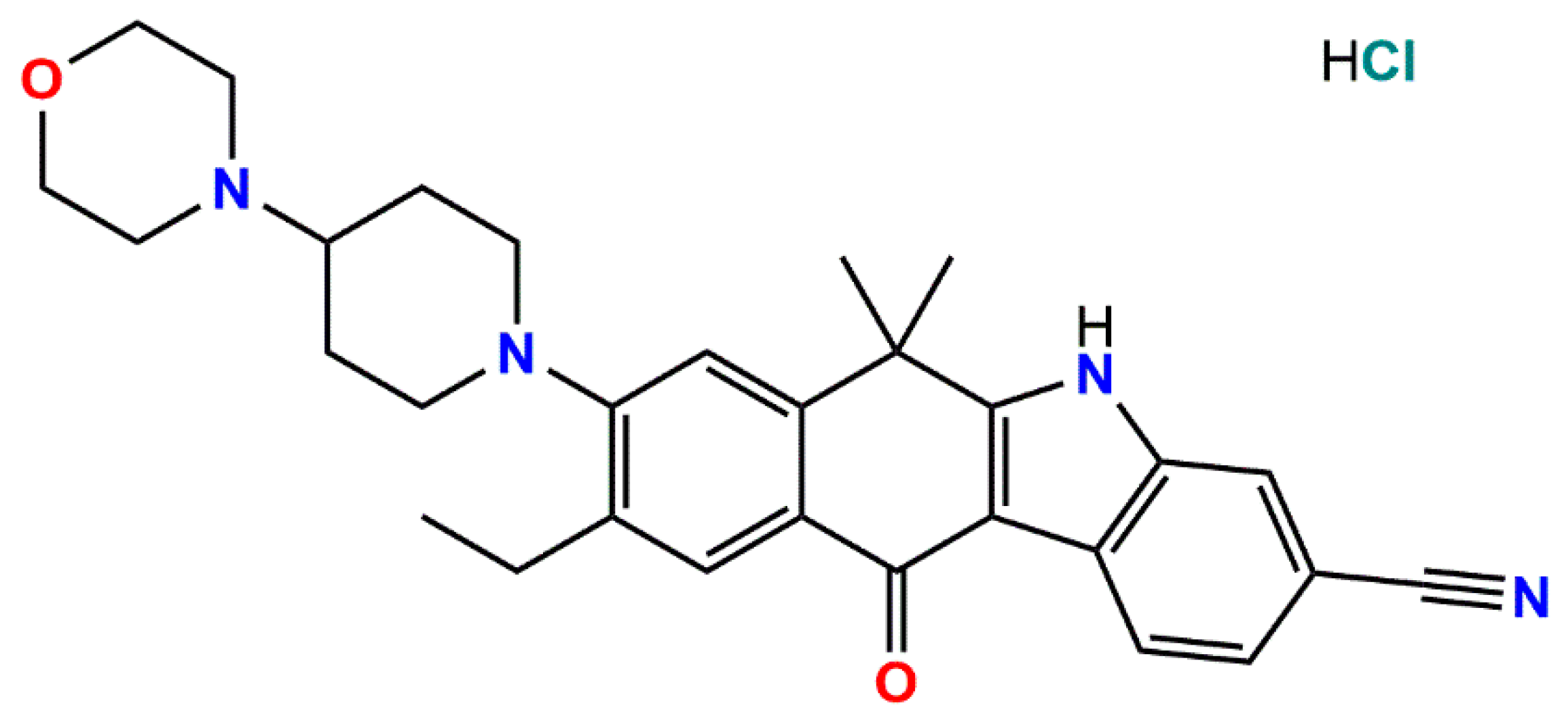
9-Ethyl-6,6-dimethyl-8-[4-(morpholin-4-yl)piperidin-1-yl]-11-oxo-6,11-dihydro-5H-benzo[b]carbazole-3-carbonitrile Hydrochloride


Abstract
1. Introduction
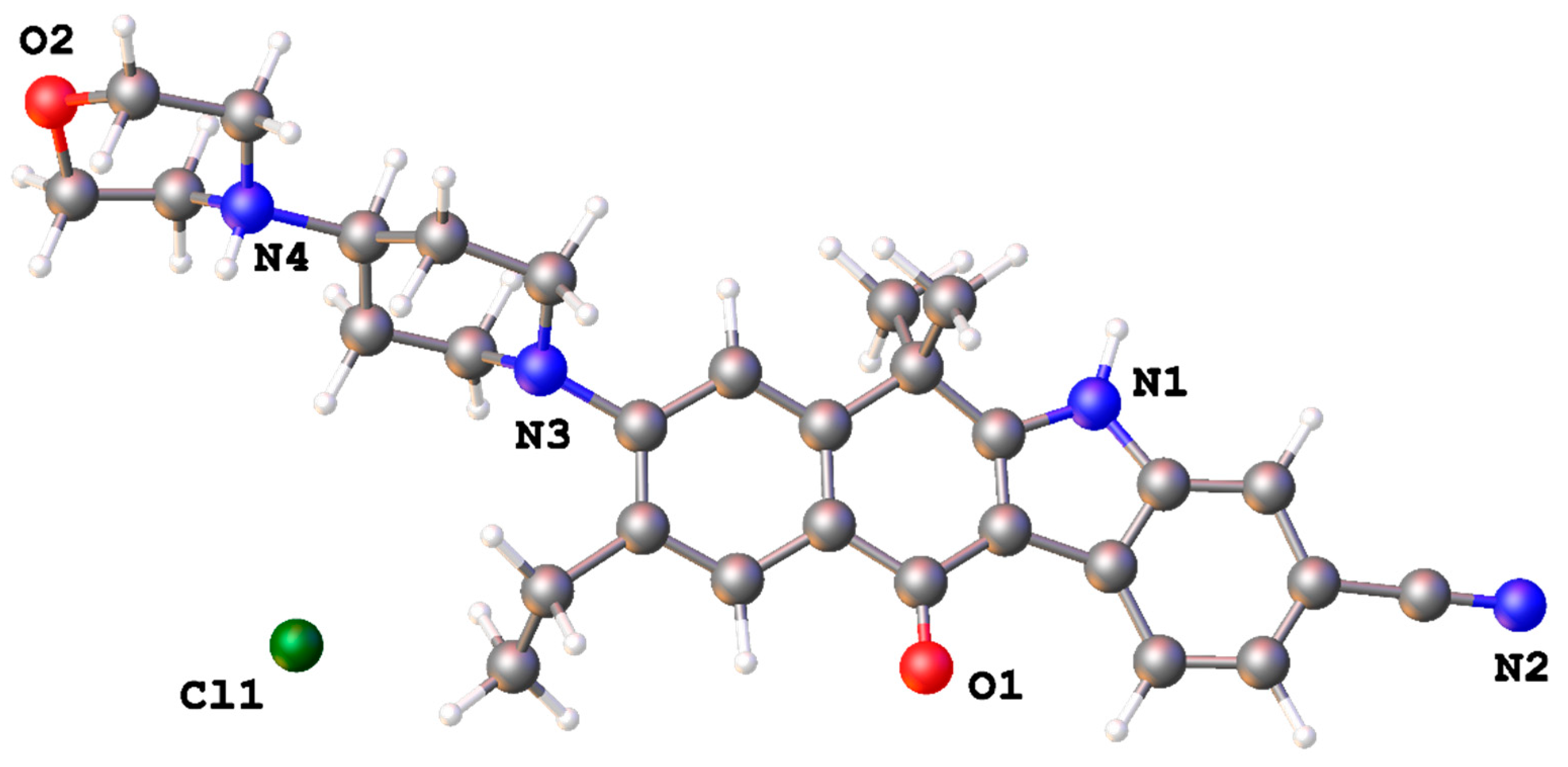
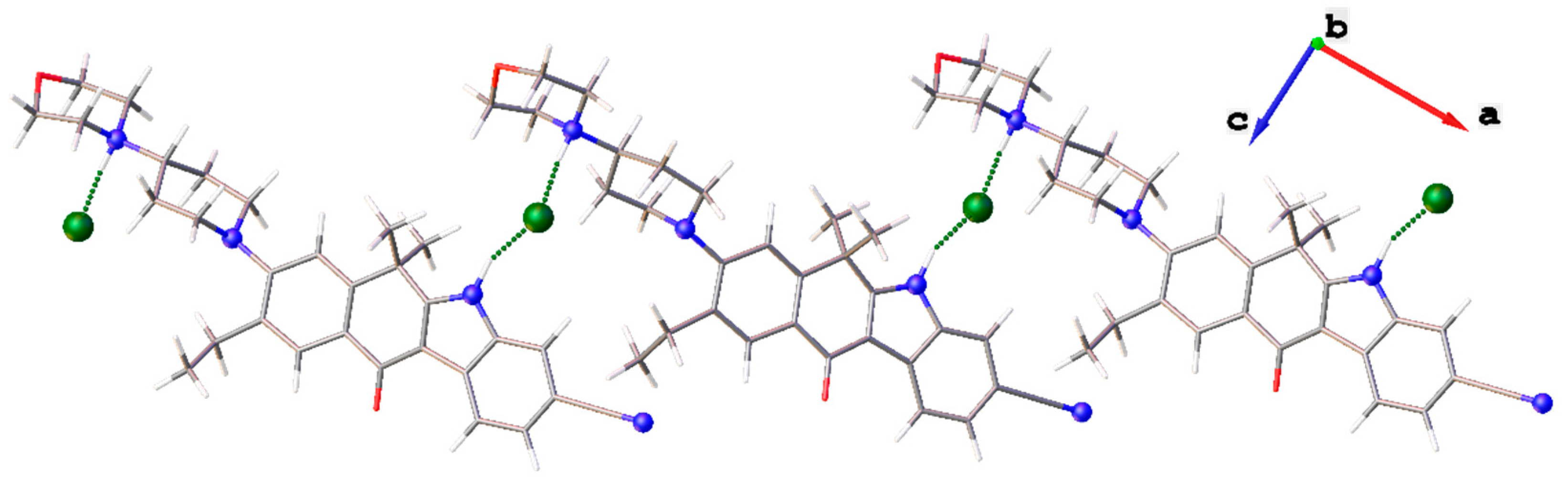
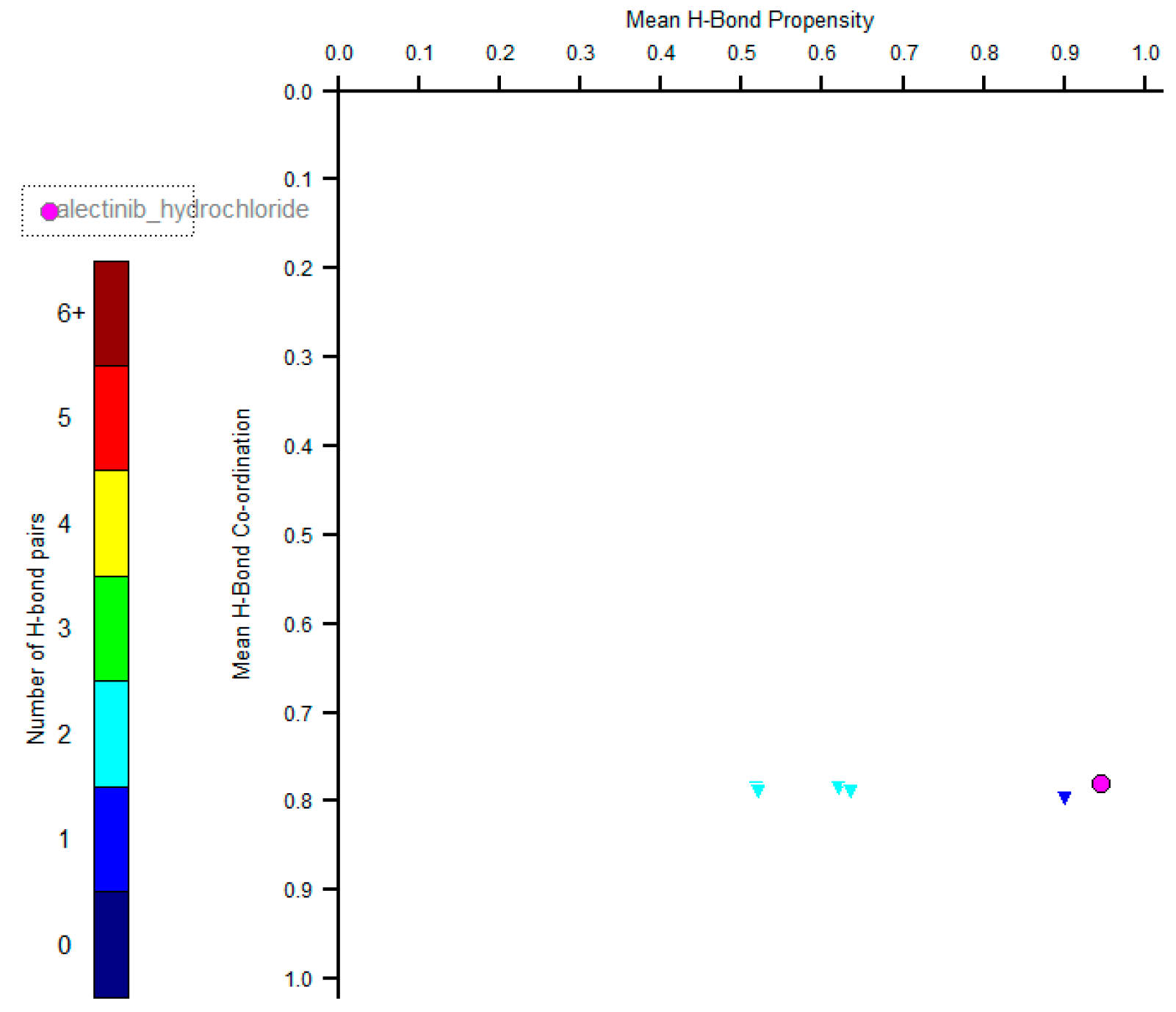
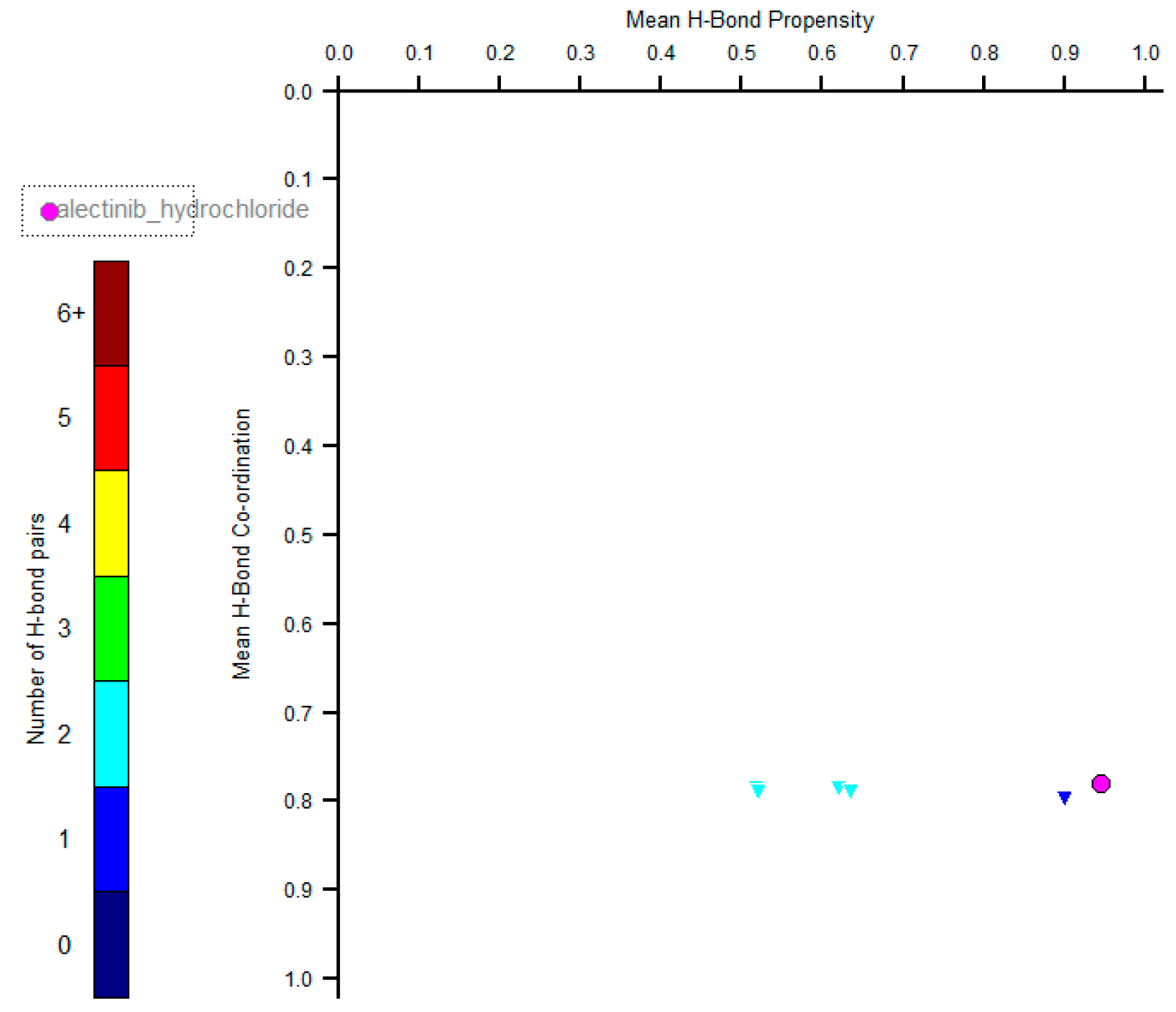
2. Results and Discussion
3. Materials and Methods
X-ray Diffraction
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McKeage, K. Alectinib: A Review of Its Use in Advanced ALK-Rearranged Non-Small Cell Lung Cancer. Drugs 2015, 75, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Ueto, T. Crystal of Tetracyclic Compound. Patent US9714229B2, 25 July 2017. Available online: https://patents.google.com/patent/US9714229B2/en?oq=alectinib++WO2015163447 (accessed on 21 December 2023).
- Kaduk, J.A.; Gates-Rector, S.; Blanton, T.N. Crystal Structure of Besifloxacin Hydrochloride, C19H22ClFN3O3Cl. Powder Diffr. 2023, 38, 43–52. [Google Scholar] [CrossRef]
- Kaduk, J.A.; Gates-Rector, S.; Blanton, T.N. Crystal Structure of Butenafine Hydrochloride, C23H28NCl. Powder Diffr. 2023, 38, 30–36. [Google Scholar] [CrossRef]
- Ens, T.M.; Kaduk, J.A.; Dosen, A.V.; Blanton, T.N. Crystal Structure of Meglumine Diatrizoate, (C7H18NO5)(C11H8I3N2O4). Powder Diffr. 2023, 38, 185–193. [Google Scholar] [CrossRef]
- Ens, T.M.; Kaduk, J.A.; Dosen, A.; Blanton, T.N. Crystal Structure of Danofloxacin Mesylate (C19H21FN3O3)(CH3O3S). Powder Diffr. 2023, 38, 194–200. [Google Scholar] [CrossRef]
- Korlyukov, A.A.; Buikin, P.A.; Dorovatovskii, P.V.; Vologzhanina, A.V. Synthesis, NoSpherA2 Refinement, and Noncovalent Bonding of Abiraterone Bromide Monohydrate. Struct. Chem. 2023, 34, 1927–1934. [Google Scholar] [CrossRef]
- Korlyukov, A.A.; Dorovatovskii, P.V.; Vologzhanina, A.V. N-(4-Methyl-3-((4-(Pyridin-3-Yl)Pyrimidin-2-Yl)Amino)Phenyl)-4-((4-Methylpiperazin-1-Yl)Methyl)Benzamide. Molbank 2022, 2022, M1461. [Google Scholar] [CrossRef]
- Volodin, A.D.; Vologzhanina, A.V.; Peresypkina, E.V.; Korlyukov, A.A. Conformational Polymorphism of Solidum Salt of Elsulfavirin. J. Struct. Chem. 2024, 65, 123238. [Google Scholar] [CrossRef]
- Coelho, A.A. TOPAS and TOPAS-Academic: An Optimization Program Integrating Computer Algebra and Crystallographic Objects Written in C++. J. Appl. Cryst. 2018, 51, 210–218. [Google Scholar] [CrossRef]
- Rietveld, H.M. A Profile Refinement Method for Nuclear and Magnetic Structures. J. Appl. Cryst. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Galek, P.T.A.; Allen, F.H.; Fábián, L.; Feeder, N. Knowledge-Based H-Bond Prediction to Aid Experimental Polymorph Screening. CrystEngComm 2009, 11, 2634–2639. [Google Scholar] [CrossRef]
- Delori, A.; Galek, P.T.A.; Pidcock, E.; Jones, W. Quantifying Homo- and Heteromolecular Hydrogen Bonds as a Guide for Adduct Formation. Chem. Eur. J. 2012, 18, 6835–6846. [Google Scholar] [CrossRef] [PubMed]
- Dmitrienko, A.O.; Bushmarinov, I.S. Reliable Structural Data from Rietveld Refinements via Restraint Consistency. J. Appl. Cryst. 2015, 48, 1777–1784. [Google Scholar] [CrossRef]
- PubChem Alectinib. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/49806720 (accessed on 29 November 2023).
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal--Amorphous-Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Compt. Mat. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D–H…A | D–H, Å | H…A, Å | D…A, Å | ∠ (DHA), ° |
|---|---|---|---|---|
| N1–H1⋯Cl1 i | 1.04 | 2.20 | 3.107(7) | 145 |
| N4–H4⋯Cl1 ii | 0.90 | 2.03 | 2.918(6) | 170 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buikin, P.A.; Vologzhanina, A.V.; Novikov, R.A.; Korlyukov, A.A. 9-Ethyl-6,6-dimethyl-8-[4-(morpholin-4-yl)piperidin-1-yl]-11-oxo-6,11-dihydro-5H-benzo[b]carbazole-3-carbonitrile Hydrochloride. Molbank 2024, 2024, M1759. https://doi.org/10.3390/M1759
Buikin PA, Vologzhanina AV, Novikov RA, Korlyukov AA. 9-Ethyl-6,6-dimethyl-8-[4-(morpholin-4-yl)piperidin-1-yl]-11-oxo-6,11-dihydro-5H-benzo[b]carbazole-3-carbonitrile Hydrochloride. Molbank. 2024; 2024(1):M1759. https://doi.org/10.3390/M1759
Chicago/Turabian StyleBuikin, Petr A., Anna V. Vologzhanina, Roman A. Novikov, and Alexander A. Korlyukov. 2024. "9-Ethyl-6,6-dimethyl-8-[4-(morpholin-4-yl)piperidin-1-yl]-11-oxo-6,11-dihydro-5H-benzo[b]carbazole-3-carbonitrile Hydrochloride" Molbank 2024, no. 1: M1759. https://doi.org/10.3390/M1759
APA StyleBuikin, P. A., Vologzhanina, A. V., Novikov, R. A., & Korlyukov, A. A. (2024). 9-Ethyl-6,6-dimethyl-8-[4-(morpholin-4-yl)piperidin-1-yl]-11-oxo-6,11-dihydro-5H-benzo[b]carbazole-3-carbonitrile Hydrochloride. Molbank, 2024(1), M1759. https://doi.org/10.3390/M1759