
(R)-(+)-3,5-Dinitro-N-(1-phenylethyl)benzothioamide


Abstract
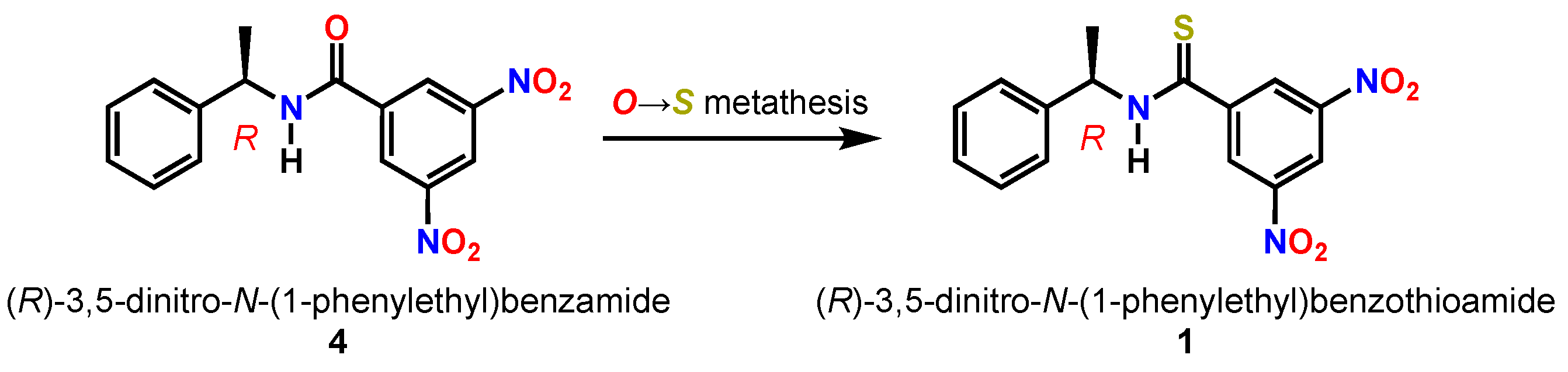
1. Introduction
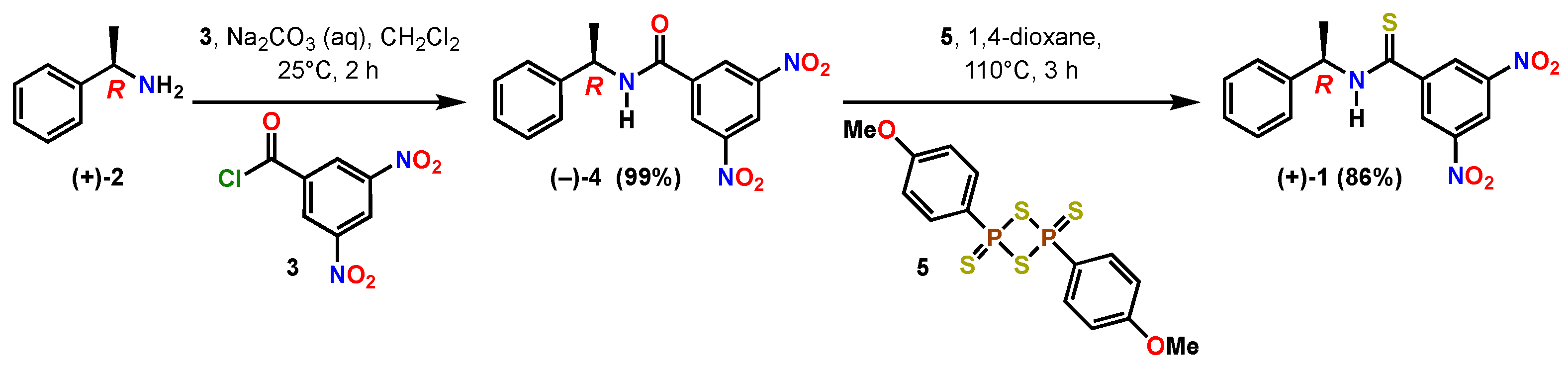
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Methods
3.3. Instrumentation and Analysis
3.4. (R)-(+)-3,5-Dinitro-N-(1-phenylethyl)benzothioamide (1)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Silva, M.S. Recent Advances in Multinuclear NMR Spectroscopy for Chiral Recognition of Organic Compounds. Molecules 2017, 22, 247. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, T.J.; Wilcox, J.D. Chiral Reagents for the Determination of Enantiomeric Excess and Absolute Configuration Using NMR Spectroscopy. Chirality 2003, 15, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Gal, J. Molecular Chirality: Language, History and Significance. In Differentiation of Enantiomers I; Springer: New York, NY, USA, 2013; pp. 1–21. [Google Scholar] [CrossRef]
- Parker, D. NMR Determination of Enantiomeric Purity. Chem. Rev. 1991, 91, 1441–1457. [Google Scholar] [CrossRef]
- Deshmukh, M.; Duñach, E.; Juge, S.; Kagan, H.B. A Convenient Family of Chiral Shift Reagents for Measurement of Enantiomeric Excesses of Sulfoxides. Tetrahedron Lett. 1984, 25, 3467–3470. [Google Scholar] [CrossRef]
- Wolf, C.; Cook, A.M.; Dannatt, J.E. Enantiodifferentiation of Multifunctional Tertiary Alcohols by NMR Spectroscopy with a Whelk-O Type Chiral Solvating Agent. Tetrahedron Asymmetry 2014, 25, 163–169. [Google Scholar] [CrossRef]
- Iwaniuk, D.P.; Wolf, C. A Versatile and Practical Solvating Agent for Enantioselective Recognition and NMR Analysis of Protected Amines. J. Org. Chem. 2010, 75, 6724–6727. [Google Scholar] [CrossRef]
- Duñach, E.; Kagan, H.B. A Simple Chiral Shift Reagent for Measurement of Enantiomeric Excesses of Phosphine Oxides. Tetrahedron Lett. 1985, 26, 2649–2652. [Google Scholar] [CrossRef]
- Pakulski, X.; Demchuk, O.M.; Kwiatosz, R.; Osinski, P.W.; Swierczynska, W.; Pietrusiewicz, K.M. The Classical Kagan’s Amides are Still Practical NMR Chiral Shift Reagents: Determination of Enantiomeric Purity of P-Chiroenic Phospholene Oxides. Tetrahedron Asymmetry 2003, 14, 1459–1462. [Google Scholar] [CrossRef]
- Kaboudin, B.; Yarahmadi, V.; Kato, J.-Y.; Yokomatsu, T. A Simple and Novel Method for the Direct Conversion of Carboxylic Acids into Thioamides. RSC Adv. 2013, 3, 6435–6441. [Google Scholar] [CrossRef]
- Lu, Z.-L.; Fun, H.-K.; Chantrapromma, S.; Brown, R.S. N-[(R)-1-Phenylethyl]thiobenzamide. Acta Cryst. 2006, E62, o1513–o1515. [Google Scholar] [CrossRef]
- Sakamoto, M.; Kawanishi, H.; Mino, T.; Kasashima, Y.; Fujita, T. Photochemical Asymmetric Synthesis of Phenyl-Bearing Quaternary Chiral Carbons Using Chiral-Memory Effect on b-Hydrogen Abstraction by Thiocarbonyl Group. Chem. Commun. 2006, 4608–4610. [Google Scholar] [CrossRef] [PubMed]
- Abboud, J.L.M.; Mo, O.; de Paz, J.L.G.; Yanez, M.; Esseffar, M.; Bouab, W.; El-Mouhtadi, M.; Mokhlisse, R.; Ballesteros, E.; Herreros, M.; et al. Thiocarbonyl versus Carbonyl Compounds: A Comparision of Intrinsic Reactivities. J. Am. Chem. Soc. 1993, 115, 12468–12476. [Google Scholar] [CrossRef]
- Madarász, A.; Dósa, Z.; Varga, S.; Soós, T.; Csámpai, A.; Pápai, I. Thiourea Derivatives as Bronsted Acid Organocatalysts. ACS Catal. 2016, 6, 4379–4387. [Google Scholar] [CrossRef]
- Rombola, M.; Sumaria, C.S.; Montgomery, T.D.; Rawal, V.H. Development of Chiral, Bifunctional Thiosquaramides: Enantioselective Michael Additions of Barbituric Acids to Nitroalkenes. J. Am. Chem. Soc. 2017, 139, 5297–5300. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.E.; Seguin, T.J.; Guan, Y.; Doney, A.C. Noncovalent Interactions in Organocatalysis and the Prospect of Computational Catalyst Desgin. Acc. Chem. Res. 2016, 49, 1061–1069. [Google Scholar] [CrossRef]
- Phillips, A.M.F.; Prechtl, M.H.G.; Pombeiro, A.J.L. Non-Covalent Interactions in Enantioselective Organocatalysis: Theoretical and Mechanistic Studies of Reactions Mediated by Dual H-Bond Donors, Bifunctional Squaramides, Thioureas and Related Catalysts. Catalysts 2021, 11, 569. [Google Scholar] [CrossRef]
- Truter, M.R. An Accurate Determination of the Crystal Structure of Thioacetamide. J. Chem. Soc. 1960, 997–1007. [Google Scholar] [CrossRef]
- Zieliński, T.; Jurczak, J. Thioamides versus Amides in Anion Binding. Tetrahedron 2005, 61, 4081–4089. [Google Scholar] [CrossRef]
- Bordwell, F.G. Equilibrium Acidities in Dimethyl Sulfoxide Solution. Acc. Chem. Res. 1988, 21, 456–463. [Google Scholar] [CrossRef]
- Lee, H.J.; Choi, Y.S.; Lee, K.B.; Park, J.; Yoon, C.J. Hydrogen Bonding Abilities of Thioamide. J. Phys. Chem. A 2002, 106, 7010–7017. [Google Scholar] [CrossRef]
- Seco, J.M.; Quiñoá, E.; Riguera, R. The Assignment of Absolute Configuration by NMR. Chem. Rev. 2004, 104, 17–118. [Google Scholar] [CrossRef]
- Yde, B.; Yousif, N.M.; Pedersen, U.; Thomsen, I.; Lawesson, S.O. Studies on Organophosphorus Compounds XLVII: Preparation of Thiated Synthons of Amides, Lactams and Imides by Use of Some New P,S-Containing Reagents. Tetrahedron 1984, 40, 2047–2052. [Google Scholar] [CrossRef]
- Ozturk, T.; Ertas, E.; Mert, O. Use of Lawesson’s Reagent in Organic Synthesis. Chem. Rev. 2007, 107, 5210–5278. [Google Scholar] [CrossRef] [PubMed]
- Spinner, E. Detection of Thiocarbonyl Groups by Infrared Spectroscopy. J. Org. Chem. 1958, 23, 2037–2038. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| 13C NMR Data 1 | 1H NMR Data 2 | |||
| Position 3 | 4 δC | 1 δC | 4 δH | 1 δH |
| a | 161.8 (s) | 191.7 (s) | - | - |
| b | 148.6 (s) | 144.5 (s) | - | - |
| c | 141.8 (s) | 148.1 (s) | - | - |
| d | 137.9 (s) | 140.4 (s) | - | - |
| e | 129.0 (d) | 129.0 (d) | 7.41–7.35 (m, 2H) | 7.43–7.29 (m, 2H) |
| f | 128.0 (d) | 128.3 (d) | 7.33–7.28 (m, 1H) | 7.43–7.29 (m, 1H) |
| g | 127.1 (d) | 126.7 (d) | 8.93 (d, J = 2.04 Hz, 2H) | 8.81 (d, J = 2.04 Hz, 2H) |
| h | 126.3 (d) | 126.8 (d) | 7.41–7.35 (m, 2H) | 7.43–7.29 (m, 2H) |
| i | 121.1 (d) | 119.9 (d) | 9.14 (t, J = 2.08 Hz, 1H) | 8.99 (t, J = 2.04 Hz, 1H) |
| j | 50.3 (d) | 56.2 (d) | 5.34 (dq, J = 7.16, 7.12 Hz, 1H) | 5.83 (dq, J = 7.20, 7.00 Hz, 1H) |
| k | 21.4 (q) | 19.9 (q) | 1.67 (d, J = 6.92 Hz, 3H) | 1.76 (d, J = 6.92 Hz, 3H) |
| l | - | - | 6.64 (s, 1H) | 8.12 (s, 1H) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donahue, M.G.; Crull, E. (R)-(+)-3,5-Dinitro-N-(1-phenylethyl)benzothioamide. Molbank 2023, 2023, M1650. https://doi.org/10.3390/M1650
Donahue MG, Crull E. (R)-(+)-3,5-Dinitro-N-(1-phenylethyl)benzothioamide. Molbank. 2023; 2023(2):M1650. https://doi.org/10.3390/M1650
Chicago/Turabian StyleDonahue, Matthew G., and Emily Crull. 2023. "(R)-(+)-3,5-Dinitro-N-(1-phenylethyl)benzothioamide" Molbank 2023, no. 2: M1650. https://doi.org/10.3390/M1650
APA StyleDonahue, M. G., & Crull, E. (2023). (R)-(+)-3,5-Dinitro-N-(1-phenylethyl)benzothioamide. Molbank, 2023(2), M1650. https://doi.org/10.3390/M1650