6-[(2S,3R)-3-(2,4-Difluorophenyl)-3-hydroxy-4-(1H-1,2,4-triazol-1-yl)butan-2-yl]-5-fluoropyrimidine-4-carbaldehyde




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
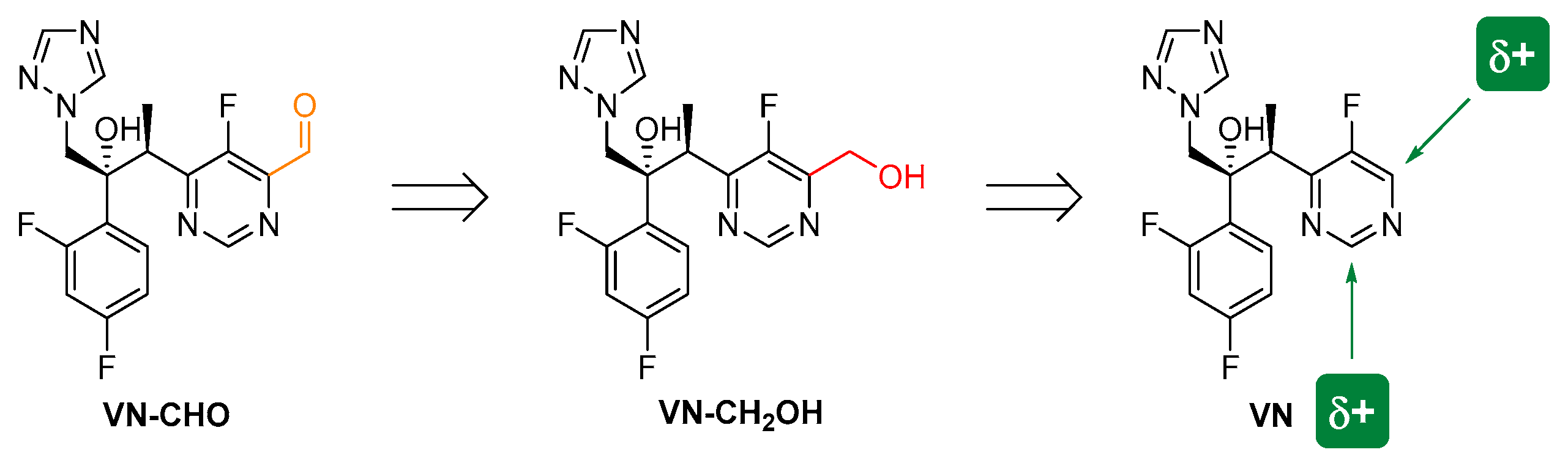
Abstract
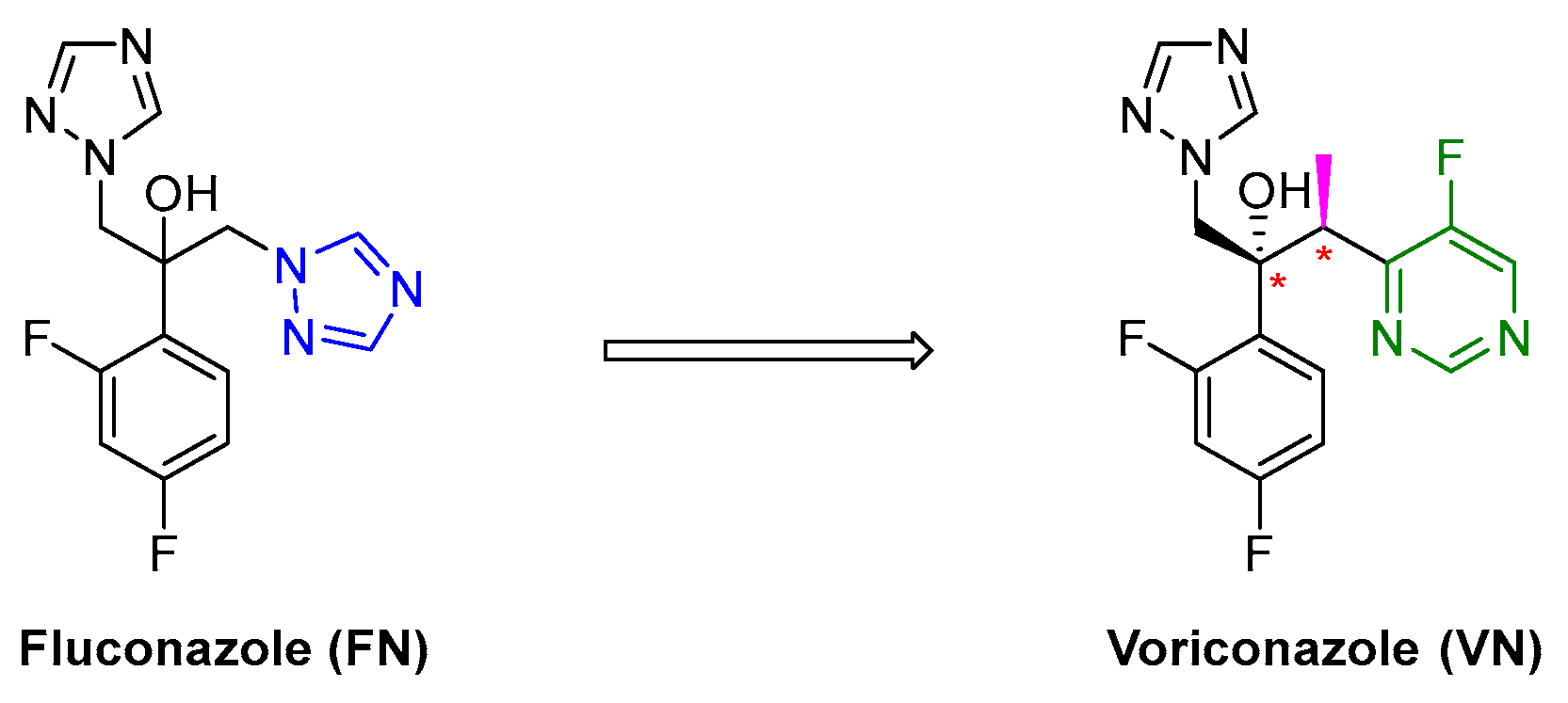
1. Introduction
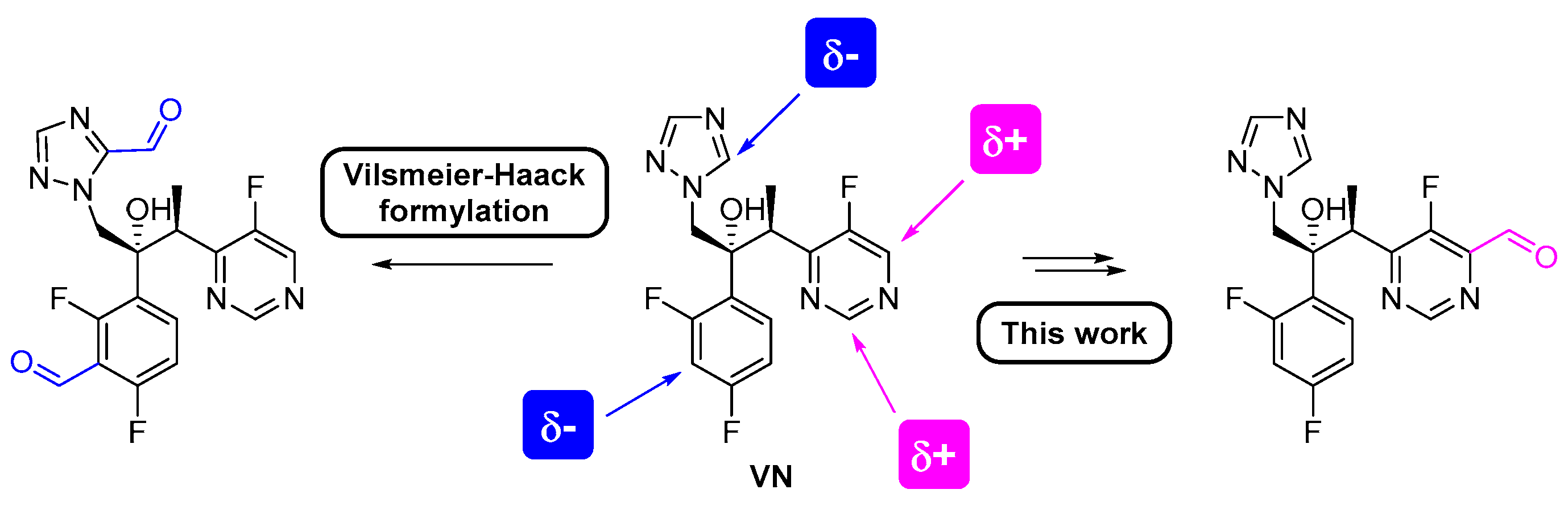
2. Results and Discussion
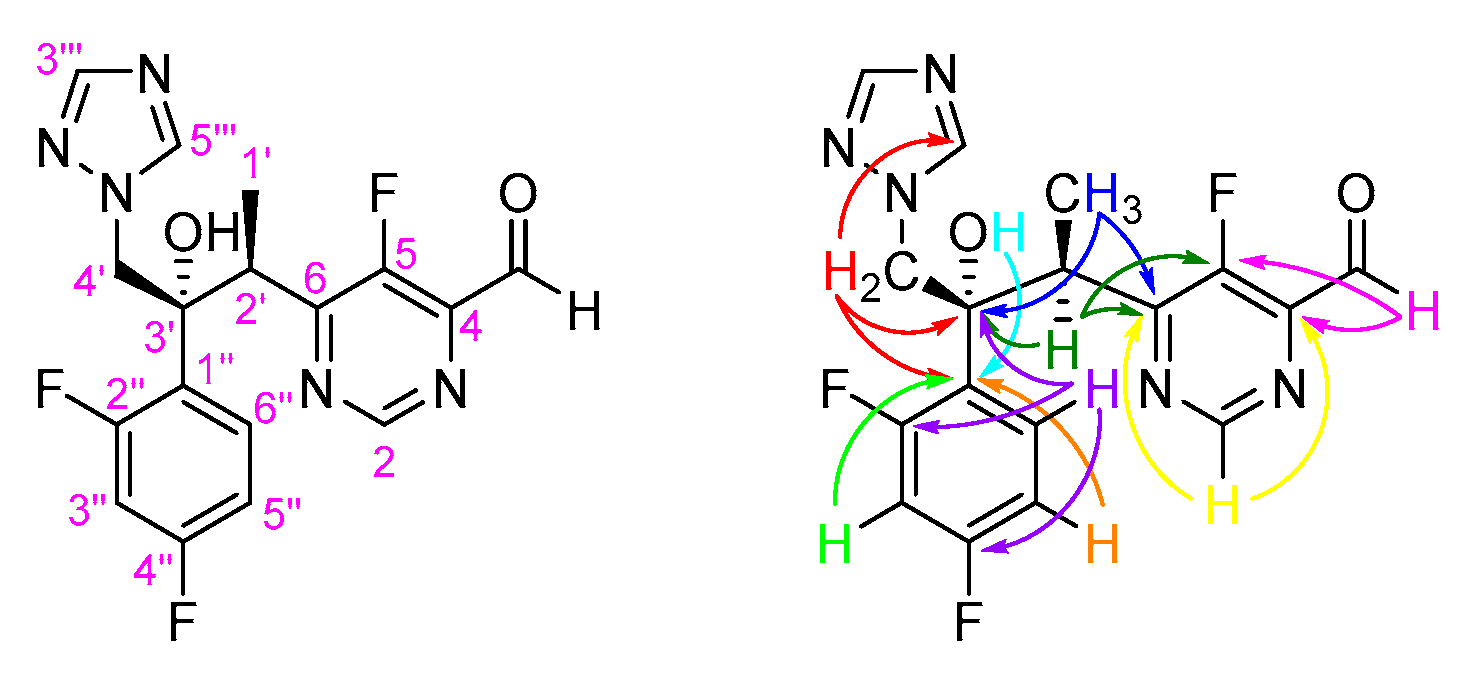
- (i)
- The doublet (JH-F 0.6 Hz) at δH 10.19 ppm, which corresponds to the resonance of the proton of the 4-CHO group.
- (ii)
- The doublet (JH-F 1.6 Hz) at δH 9.08 ppm, which corresponds to the resonance of H-2 of the pyrimidine ring.
- (iii)
- Two singlets at δH 7.54 and 7.96 ppm, which correspond to the resonance of H-3′′′ and H-5′′′ of the 1,2,4-triazole moiety.
- (iv)
- The broad singlet at δH 6.19 ppm, which corresponds to the resonance of the proton of the 3′-OH group.
- (v)
- The doublet (J1′-2′ 7.1 Hz) at δH 1.15 ppm, which corresponds to the resonance of the protons of the 2′-CH3 group.
- (i)
- C-4 (δc 144.2 ppm) as a doublet (2J4-F 7.0 Hz) due to its connectivities with 4-CHO and H-2.
- (ii)
- C-5 (δc 153.8 ppm) as a doublet (1J5-F 282.3 Hz) due to its connectivities with 4-CHO and H-2′.
- (iii)
- C-6 (δc 163.6 ppm) as a doublet (2J6-F 13.3 Hz) due to its connectivities with H-2, H-2′ and 2′-CH3.
- (iv)
- C-3′ (δc 77.6 ppm) due to its connectivities with 2′-CH3, H-2′, H-4′ and H-6′′.
- (v)
- C-1′′ (δc 123.4 ppm) as a doublet of doublets (4J1′′-F 4.0 and 2J1′′-F 12.3 Hz) due to its connectivities with 3′-OH, H-4′, H-3′′ and H-5′′.
- (vi)
- C-2′′ and C-4′′ (δc 156.7–164.6 ppm) due to its connectivities with H-6′′. However, their signals are hidden by the background noise in the 13C NMR spectrum, which prevented their accurate assignment.
3. Materials and Methods
3.1. General Remarks
3.2. General Procedure for the Synthesis of 6-[(2S,3R)-3-(2,4-Difluorophenyl)-3-hydroxy-4-(1H-1,2,4-triazol-1-yl)butan-2-yl]-5-fluoropyrimidine-4-carbaldehyde (VN-CHO)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arendrup, M.C. Update on antifungal resistance in Aspergillus and Candida. Clin. Microbiol. Infect. 2014, 20, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Ni, T.; Xie, F.; Hao, Y.; Yu, S.; Chai, X.; Jin, Y.; Wang, T.; Jiang, Y.; Zhang, D. Design, synthesis, and structure-activity relationship studies of novel triazole agents with strong antifungal activity against Aspergillus fumigatus. Bioorg. Med. Chem. Lett. 2020, 30, 126951. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Effron, G. Molecular Markers of Antifungal Resistance: Potential Uses in Routine Practice and Future Perspectives. J. Fungi 2021, 7, 197. [Google Scholar] [CrossRef] [PubMed]
- Shafiei, M.; Peyton, L.; Hashemzadeh, M.; Foroumadi, A. History of the development of antifungal azoles: A review on structures, SAR, and mechanism of action. Bioorg. Chem. 2020, 104, 104240. [Google Scholar] [CrossRef] [PubMed]
- Ghobadi, E.; Saednia, S.; Emami, S. Synthetic approaches and structural diversity of triazolylbutanols derived from voriconazole in the antifungal drug development. Eur. J. Med. Chem. 2022, 231, 114161. [Google Scholar] [CrossRef] [PubMed]
- Butters, M.; Ebbs, J.; Green, S.P.; MacRae, J.; Morland, M.C.; Murtiashaw, C.W.; Pettman, A.J. Process Development of Voriconazole: A Novel Broad-Spectrum Triazole Antifungal Agent. Org. Process Res. Dev. 2001, 5, 28–36. [Google Scholar] [CrossRef]
- Sundaram, D.T.S.S.; Mitra, J.; Islam, A.; Prabahar, K.J.; Venkateswara Rao, B.; Paul Douglas, S. Synthesis of Isomeric and Potent Impurities of the Triazole-Based Antifungal Drug Voriconazole. Sci. Pharm. 2015, 83, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Moir, M.; Danon, J.J.; Reekie, T.A.; Kassiou, M. An overview of late-stage functionalization in today’s drug discovery. Expert Opin. Drug Discov. 2019, 14, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Cernak, T.; Dykstra, K.D.; Tyagarajan, S.; Vachal, P.; Krska, S.W. The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 2016, 45, 546–576. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.H.; Twilton, J.; MacMillan, D.W.C. Photoredox Catalysis in Organic Chemistry. J. Org. Chem. 2016, 81, 6898–6926. [Google Scholar] [CrossRef]
- Huff, C.A.; Cohen, R.D.; Dykstra, K.D.; Streckfuss, E.; DiRocco, D.A.; Krska, S.W. Photoredox-Catalyzed Hydroxymethylation of Heteroaromatic Bases. J. Org. Chem. 2016, 81, 6980–6987. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa, J.L.C.; Albuquerque, H.M.T.; Silva, A.M.S. 6-[(2S,3R)-3-(2,4-Difluorophenyl)-3-hydroxy-4-(1H-1,2,4-triazol-1-yl)butan-2-yl]-5-fluoropyrimidine-4-carbaldehyde. Molbank 2023, 2023, M1603. https://doi.org/10.3390/M1603
Sousa JLC, Albuquerque HMT, Silva AMS. 6-[(2S,3R)-3-(2,4-Difluorophenyl)-3-hydroxy-4-(1H-1,2,4-triazol-1-yl)butan-2-yl]-5-fluoropyrimidine-4-carbaldehyde. Molbank. 2023; 2023(1):M1603. https://doi.org/10.3390/M1603
Chicago/Turabian StyleSousa, Joana L. C., Hélio M. T. Albuquerque, and Artur M. S. Silva. 2023. "6-[(2S,3R)-3-(2,4-Difluorophenyl)-3-hydroxy-4-(1H-1,2,4-triazol-1-yl)butan-2-yl]-5-fluoropyrimidine-4-carbaldehyde" Molbank 2023, no. 1: M1603. https://doi.org/10.3390/M1603
APA StyleSousa, J. L. C., Albuquerque, H. M. T., & Silva, A. M. S. (2023). 6-[(2S,3R)-3-(2,4-Difluorophenyl)-3-hydroxy-4-(1H-1,2,4-triazol-1-yl)butan-2-yl]-5-fluoropyrimidine-4-carbaldehyde. Molbank, 2023(1), M1603. https://doi.org/10.3390/M1603