
Synthesis of 2-[2-(Ethoxymethoxy)phenyl]spiro[cyclopropane-1,2′-indene]-1′,3′-dione

 , ,
, , 
Abstract
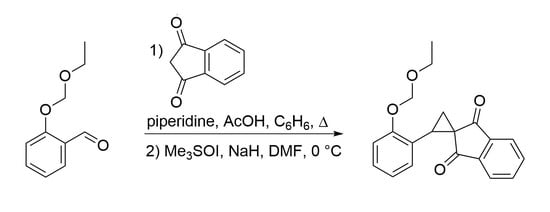
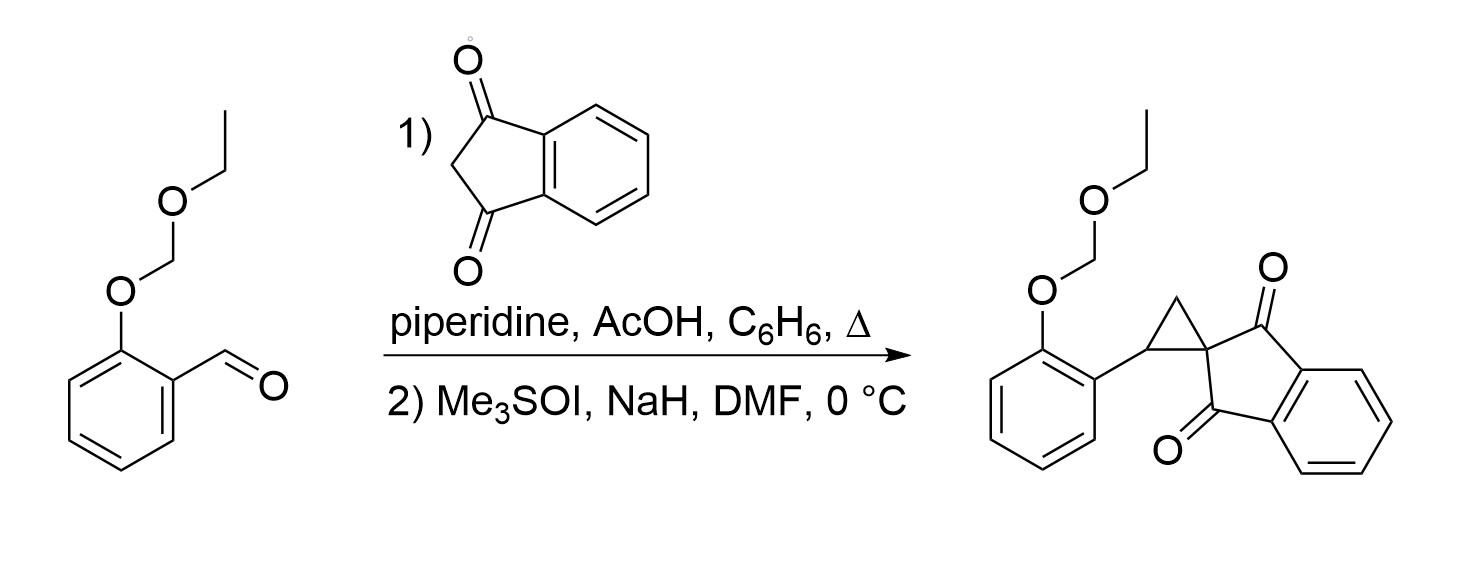{kind=link}
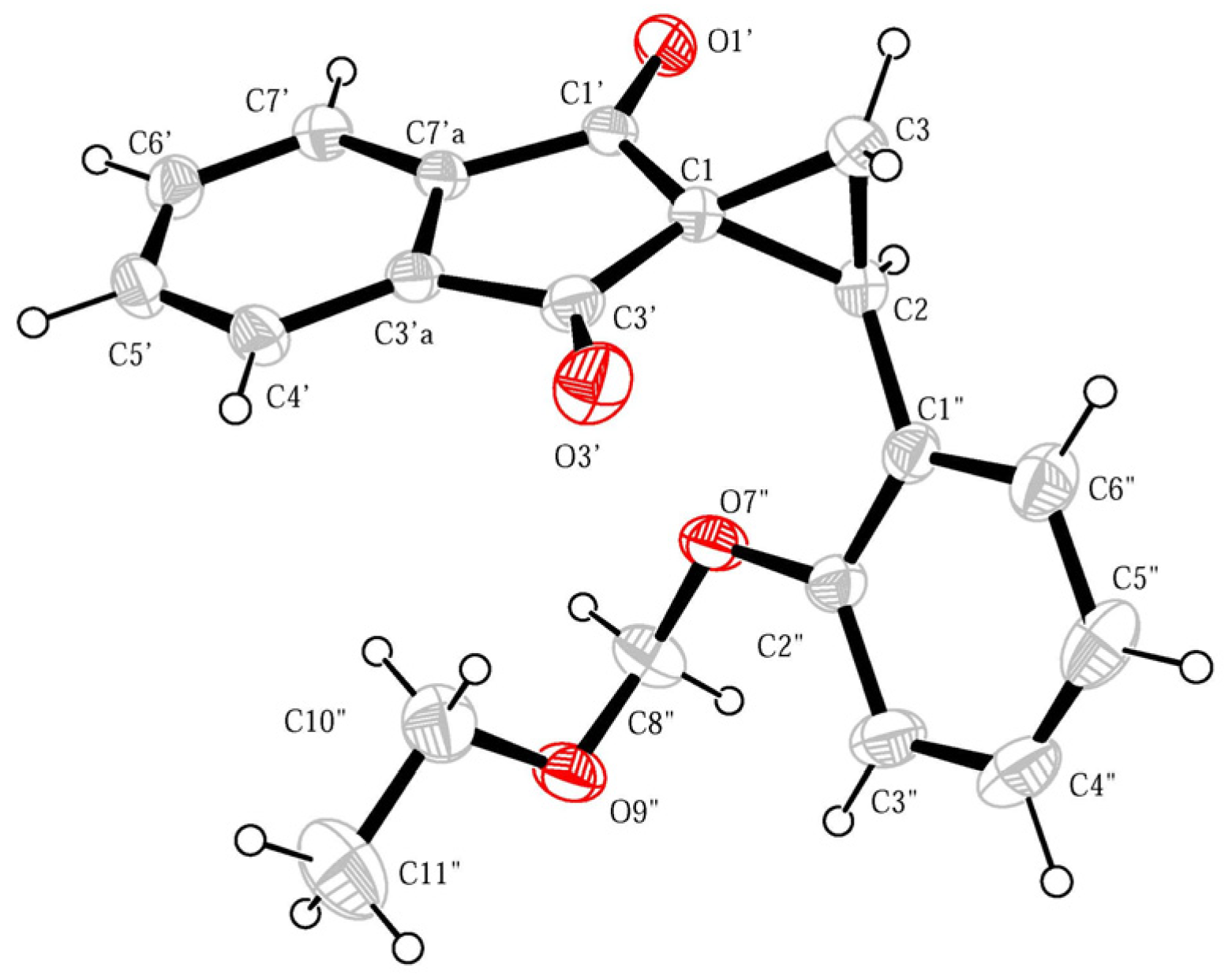{kind=link}
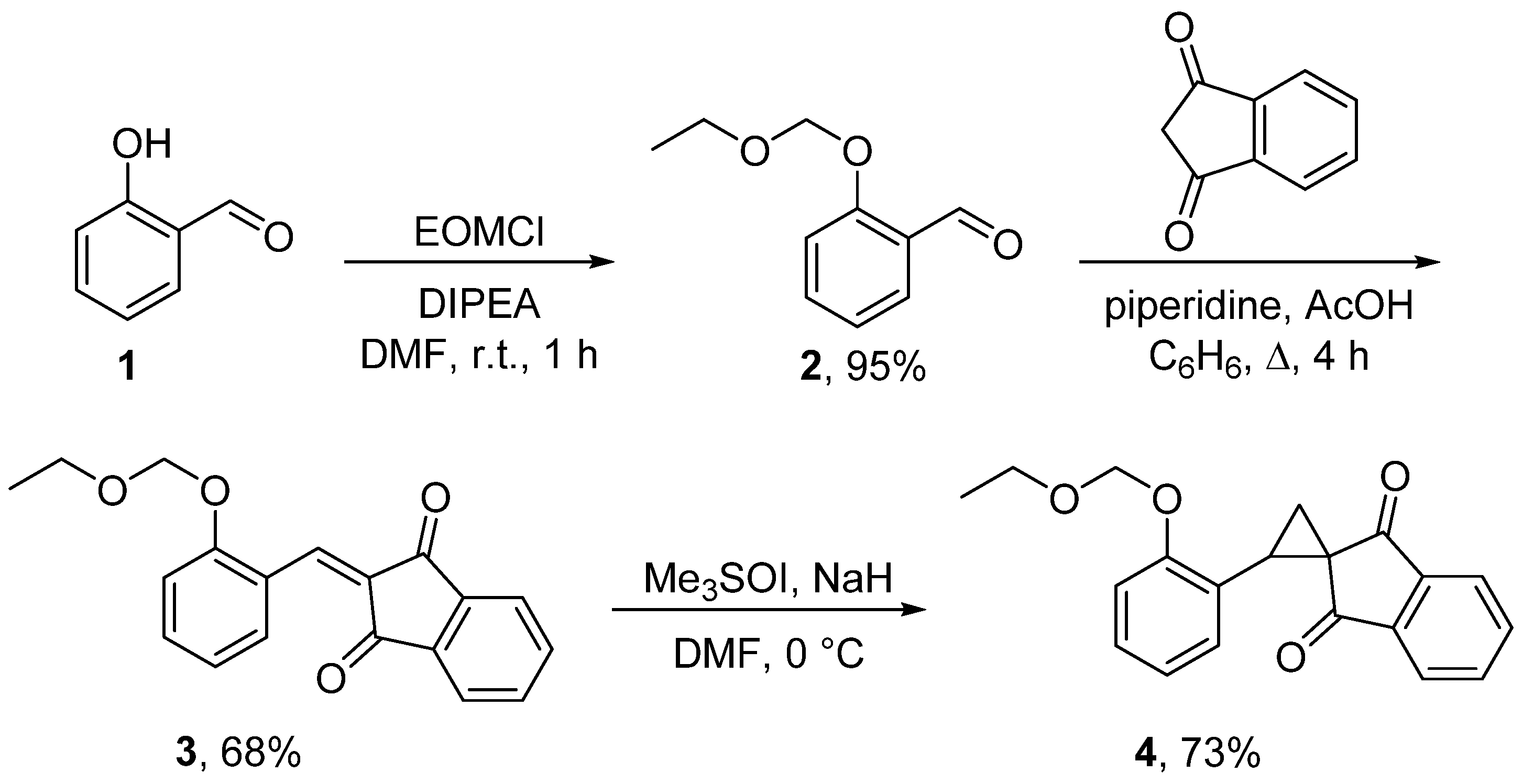{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. 2-[2-(Ethoxymethoxy)benzylidene]indene-1,3(2H)-dione (3)
3.2. 2-[2-(Ethoxymethoxy)phenyl]spiro[cyclopropane-1,2′-indene]-1′,3′-dione (4)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Augustin, A.U.; Werz, D.B. Exploiting Heavier Organochalcogen Compounds in Donor−Acceptor Cyclopropane Chemistry. Acc. Chem. Res. 2021, 54, 1528–1541. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, K.; Das, S. Recent advances in ring-opening of donor acceptor cyclopropanes using C-nucleophiles. Org. Biomol. Chem. 2021, 19, 965–982. [Google Scholar] [CrossRef]
- Pirenne, V.; Muriel, B.; Waser, J. Catalytic Enantioselective Ring-Opening Reactions of Cyclopropanes. Chem. Rev. 2021, 121, 227–263. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Varshnaya, R.K.; Dey, R.; Banerjee, P. Donor−Acceptor Cyclopropanes as an Expedient Building Block Towards the Construction of Nitrogen-Containing Molecules: An Update. Adv. Synth. Catal. 2020, 362, 1447–1484. [Google Scholar] [CrossRef]
- Ivanova, O.A.; Trushkov, I.V. Donor−Acceptor Cyclopropanes in the Synthesis of Carbocycles. Chem. Rec. 2019, 19, 2189–2208. [Google Scholar] [CrossRef] [PubMed]
- Tomilov, Y.V.; Menchikov, L.G.; Novikov, R.A.; Ivanova, O.A.; Trushkov, I.V. Methods for the synthesis of donor–acceptor cyclopropanes. Russ. Chem. Rev. 2018, 87, 201–250. [Google Scholar] [CrossRef]
- Schneider, T.F.; Kaschel, J.; Werz, D.B. A New Golden Age for Donor-Acceptor Cyclopropanes. Angew. Chem. Int. Ed. 2014, 53, 5504–5523. [Google Scholar] [CrossRef]
- Belaya, M.A.; Knyazev, D.A.; Borisov, D.D.; Novikov, R.A.; Tomilov, Y.V. GaCl3-Mediated Cascade [2 + 4]-Cycloaddition/[4 + 2]-Annulation of Donor–Acceptor Cyclopropanes with Conjugated Dienes: Strategy for the Construction of Benzobicyclo[3.3.1]nonane Skeleton. J. Org. Chem. 2021, 86, 8089–8100. [Google Scholar] [CrossRef]
- Belaya, M.A.; Knyazev, D.A.; Novikov, R.A.; Tomilov, Y.V. “Diels-Alder reaction” in the ionic version: GaCl3-promoted formation of substituted cyclohexenes from donor–acceptor cyclopropanes and dienes. Tetrahedron Lett. 2020, 61, 151990. [Google Scholar] [CrossRef]
- Borisov, D.D.; Novikov, R.A.; Tomilov, Y.V. GaCl3-Mediated Reactions of Donor–Acceptor Cyclopropanes with Aromatic Aldehydes. Angew. Chem. Int. Ed. 2016, 55, 12233–12237. [Google Scholar] [CrossRef]
- Novikov, R.A.; Tarasova, A.V.; Korolev, V.A.; Timofeev, V.P.; Tomilov, Y.V. A New Type of Donor–Acceptor Cyclopropane Reactivity: The Generation of Formal 1,2- and 1,4-Dipoles. Angew. Chem. Int. Ed. 2014, 53, 3187–3191. [Google Scholar] [CrossRef] [PubMed]
- Novikov, R.A.; Korolev, V.A.; Timofeev, V.P.; Tomilov, Y.V. New dimerization and cascade oligomerization reactions of dimethyl 2-phenylcyclopropan-1,1-dicarboxylate catalyzed by Lewis acids. Tetrahedron Lett. 2011, 52, 4996–4999. [Google Scholar] [CrossRef]
- Fadeev, A.A.; Makarov, A.S.; Ivanova, O.A.; Uchuskin, M.G.; Trushkov, I.V. Extended Corey-Chaykovsky reactions: Transformations of 2-hydroxychalcones to benzannulated 2,8-dioxabicyclo[3.2.1]octanes and 2,3-dihydrobenzofurans. Org. Chem. Front. 2022, 9, 737–744. [Google Scholar] [CrossRef]
- Ivanova, O.A.; Andronov, V.A.; Vasin, V.S.; Shumsky, A.N.; Rybakov, V.B.; Voskressensky, L.G.; Trushkov, I.V. Expanding the Reactivity of Donor–Acceptor Cyclopropanes: Synthesis of Benzannulated Five-Membered Heterocycles via Intramolecular Attack of a Pendant Nucleophilic Group. Org. Lett. 2018, 20, 7947–7952. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, K.L.; Bezzubov, S.I.; Melnikov, M.Y.; Budynina, E.M. Donor–acceptor cyclopropanes as ortho-quinone methide equivalents in formal (4+2)-cycloaddition to alkenes. Org. Biomol. Chem. 2018, 16, 3897–3909. [Google Scholar] [CrossRef]
- Xiao, J.-A.; Peng, H.; Liang, J.-S.; Meng, R.-F.; Su, W.; Xiao, Q.; Yang, H. Gold/scandium bimetallic relay catalysis of formal [5+2]- and [4+2]-annulations: Access to tetracyclic indole scaffolds. Chem. Commun. 2021, 57, 13369–13372. [Google Scholar] [CrossRef]
- Unnava, R.; Chahal, K.; Reddy, K.R. Synthesis of substituted 1,2-dihydroisoquinolines via Ni(II) and Cu(I)/Ag(I) catalyzed double nucleophilic addition of arylamines to ortho-alkynyl donor–acceptor cyclopropanes (o-ADACs). Org. Biomol. Chem. 2021, 19, 6025–6029. [Google Scholar] [CrossRef]
- Wang, D.; Zhao, J.; Chen, J.; Xu, Q.; Li, H. Intramolecular Arylative Ring Opening of Donor–Acceptor Cyclopropanes in the Presence of Triflic Acid: Synthesis of 9H-Fluorenes and 9,10-Dihydrophenanthrenes. Asian J. Org. Chem. 2019, 8, 2032–2036. [Google Scholar] [CrossRef]
- Mikhaylov, A.A.; Dilman, A.D.; Novikov, R.A.; Khoroshutina, Y.A.; Struchkova, M.I.; Arkhipov, D.E.; Nelyubina, Y.V.; Tabolin, A.A.; Ioffe, S.L. Tandem Pd-catalyzed C–C coupling/recyclization of 2-(2-bromoaryl)cyclopropane-1,1-dicarboxylates with primary nitro alkanes. Tetrahedron Lett. 2016, 57, 11–14. [Google Scholar] [CrossRef]
- Ma, W.; Fang, J.; Ren, J.; Wang, Z. Lewis Acid Catalyzed Formal Intramolecular [3 + 3] Cross-Cycloaddition of Cyclopropane 1,1-Diesters for Construction of Benzobicyclo[2.2.2]octane Skeletons. Org. Lett. 2015, 17, 4180–4183. [Google Scholar] [CrossRef]
- Wang, L.-F.; Shi, Z.-F.; Cao, X.-P.; Li, B.-S.; An, P. Construction of fused- and spiro-oxa-[n.2.1] skeletons by a tandem epoxide rearrangement/intramolecular [3+2] cycloaddition of cyclopropanes with carbonyls. Chem. Commun. 2014, 50, 8061–8064. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Ren, J.; Wang, Z. Acid-Catalyzed Domino Meinwald Rearrangement of Epoxides/Intramolecular [3+2] Cross-Cycloaddition of Cyclopropane-1,1-dicarboxylates. Eur. J. Org. Chem. 2014, 2014, 3561–3564. [Google Scholar] [CrossRef]
- Flisar, M.E.; Emmett, M.R.; Kerr, M.A. Catalyst-Free Tandem Ring-Opening/Click Reaction of Acetylene-Bearing Donor–Acceptor Cyclopropanes. Synlett 2014, 25, 2297–2300. [Google Scholar] [CrossRef]
- Wang, Z. Polar Intramolecular Cross-Cycloadditions of Cyclopropanes toward Natural Product Synthesis. Synlett 2012, 23, 2311–2327. [Google Scholar] [CrossRef]
- Xia, X.-F.; Song, X.-R.; Liu, X.-Y.; Liang, Y.-M. Lewis Acid-Catalyzed Intramolecular [3+2] Cycloaddition of Cyclopropane 1,1-Diesters with Alkynes for the Synthesis of Cyclopenta[c]chromene Skeletons. Chem. Asian J. 2012, 7, 1538–1541. [Google Scholar] [CrossRef]
- Chagarovskiy, A.O.; Strel’tsova, E.D.; Rybakov, V.B.; Levina, I.I.; Trushkov, I.V. Synthesis of 2,3-diaryl-2,3,4,4a-tetrahydro-5H-indeno[1,2-c]pyridazine-5-ones. Chem. Heterocycl. Compd. 2019, 55, 240–245. [Google Scholar] [CrossRef]
- Qian, P.; Du, B.; Song, R.; Wu, X.; Mei, H.; Han, J.; Pan, Y. N-Iodosuccinimide-Initiated Spirocyclopropanation of Styrenes with 1,3-Dicarbonyl Compound for the Synthesis of Spirocyclopropanes. J. Org. Chem. 2016, 81, 6546–6553. [Google Scholar] [CrossRef]
- Nambu, H.; Fukumoto, M.; Hirota, W.; Ono, N.; Yakura, T. An efficient synthesis of cycloalkane-1,3-dione-2-spirocyclopropanes from 1,3-cycloalkanediones using (1-aryl-2-bromoethyl)dimethylsulfonium bromides: Application to a one-pot synthesis of tetrahydroindol-4(5H)-one. Tetrahedron Lett. 2015, 56, 4312–4315. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanova, O.A.; Shorokhov, V.V.; Andreev, I.A.; Ratmanova, N.K.; Rybakov, V.B.; Strel’tsova, E.D.; Trushkov, I.V. Synthesis of 2-[2-(Ethoxymethoxy)phenyl]spiro[cyclopropane-1,2′-indene]-1′,3′-dione. Molbank 2023, 2023, M1604. https://doi.org/10.3390/M1604
Ivanova OA, Shorokhov VV, Andreev IA, Ratmanova NK, Rybakov VB, Strel’tsova ED, Trushkov IV. Synthesis of 2-[2-(Ethoxymethoxy)phenyl]spiro[cyclopropane-1,2′-indene]-1′,3′-dione. Molbank. 2023; 2023(1):M1604. https://doi.org/10.3390/M1604
Chicago/Turabian StyleIvanova, Olga A., Vitaly V. Shorokhov, Ivan A. Andreev, Nina K. Ratmanova, Victor B. Rybakov, Elena D. Strel’tsova, and Igor V. Trushkov. 2023. "Synthesis of 2-[2-(Ethoxymethoxy)phenyl]spiro[cyclopropane-1,2′-indene]-1′,3′-dione" Molbank 2023, no. 1: M1604. https://doi.org/10.3390/M1604
APA StyleIvanova, O. A., Shorokhov, V. V., Andreev, I. A., Ratmanova, N. K., Rybakov, V. B., Strel’tsova, E. D., & Trushkov, I. V. (2023). Synthesis of 2-[2-(Ethoxymethoxy)phenyl]spiro[cyclopropane-1,2′-indene]-1′,3′-dione. Molbank, 2023(1), M1604. https://doi.org/10.3390/M1604