2. Results and Discussion
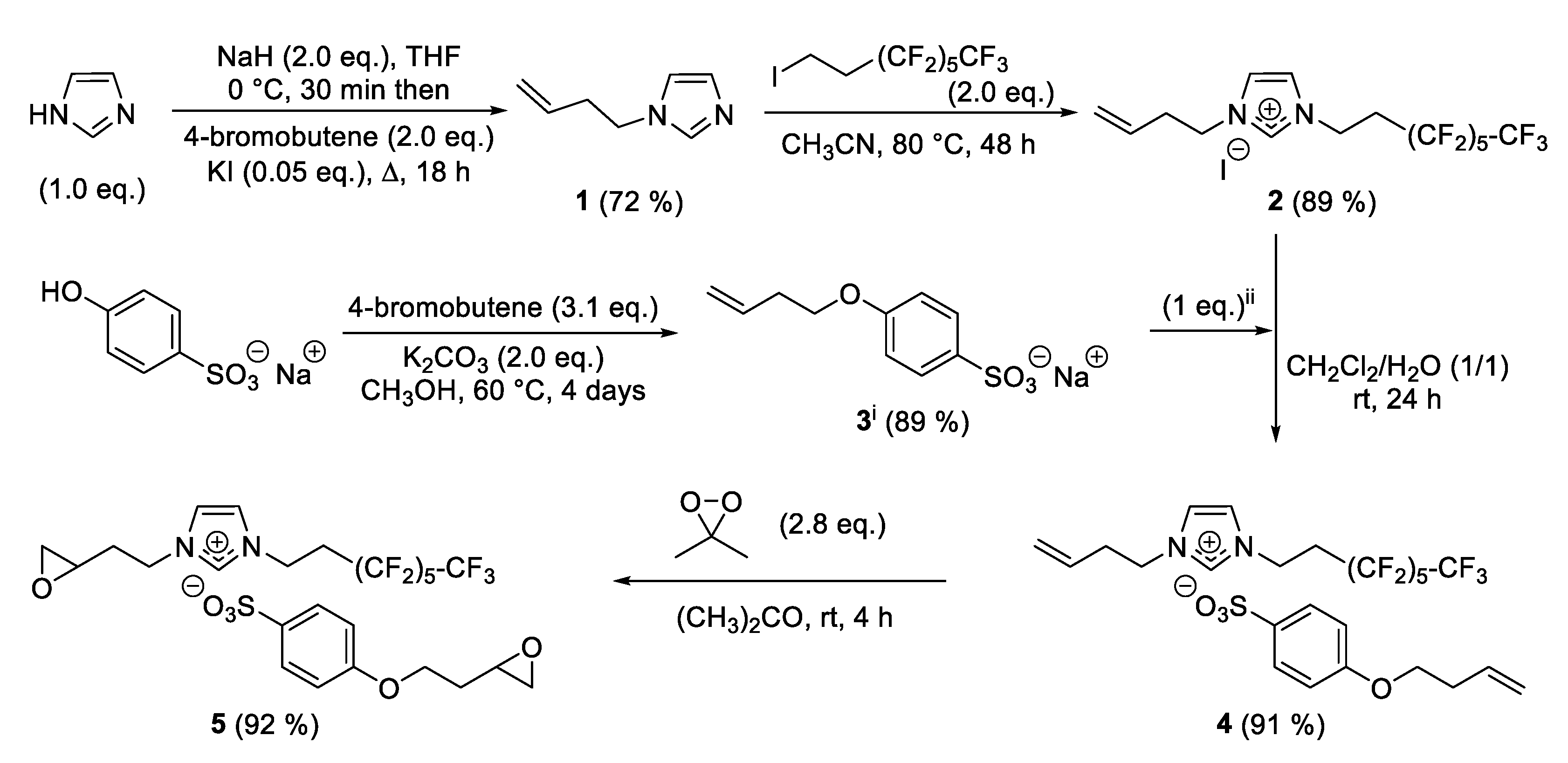
Although 1-vinylimidazole is a commercially available reagent, we opted for the synthesis of 1-(3-buten-1-yl)imidazole
1, which makes more sense in this sequence with a more oxidizable alkene. Thus, imidazolium iodide
2 was synthesized by two consecutive nucleophilic substitutions of the imidazole. The first alkylation required sodium hydride to improve the reactivity of the heterocycle and a catalytic amount of iodide potassium to achieve an efficient reaction with 4-bromo-1-butene. After 18 h at reflux, 1-(3-buten-1-yl)imidazole
1 was isolated in a 72% yield—this reaction can be carried out over several grams did not require purification. For the second step, we started from a stoichiometric amount of the previous
N-alkylated imidazole and 1H,1H,2H,2H-perfluooctyl iodide at reflux of acetonitrile. This quaternization reaction was followed by
1H-NMR and TLC (see part IVa of the
Supplementary Materials) affording 71% of salt after 24 h without significant improvement after 48 h. Thereafter, this alkylation was performed with 2 equivalents of perfluooctyl iodide to reach 89% of imidazolium
2 after 48 h at 80 °C (
Scheme 1). After reaction, the unreacted starting material was eliminated by two successive treatments with hydrochloric acid (10%). This fluorinated salt
2 behaved as a surfactant during washing (see part IVb of the
Supplementary Materials). Depending on the NMR result, flash chromatography on silica gel can be used as a second complementary method to isolate pure imidazolium
2.
At the same time, sodium 4-hydroxybenzenesulfonate was used as a widely available and inexpensive starting material of functionalized sulfonate anions. Once again, we used 4-bromo-1-butene to generate a particularly interesting terminal alkene in the anionic part of the salt. After optimization, an excess of alkylating reagent was required in the presence of potassium carbonate to give sulfonate
3 in 89% yield after 24 h in methanol. To exchange iodide of imidazolium
2, an anionic metathesis was carried out in a mixture dichloromethane/water (1/1) using a stoichiometric amount of sulfonate
3 previously synthesized. After 24 h at room temperature, imidazolium sulfonate
4 was mainly observed with a conversion of 59%. This anion exchange was slightly improved by using two equivalents of
3 to reach 70% under similar conditions as the previous attempt. This optimization confirmed the advantage of using an excess of sulfonate
3 but in a two-step addition by stirring a stoichiometric mixture of homemade sulfonate
3 and imidazolium
4 for 24 h before adding one more equivalent of sulfonate to generate a complete exchange of the iodide (see
Section 3.5). Under these optimized conditions, several extractions with dichloromethane provided the pure perfluorinated salt with an isolated yield of 91%.
This new ionic liquid
4 was oxidized in the presence of freshly prepared dimethyldioxirane (2.8 eq.) in acetone [
12]. The reaction mixture was stirred at room temperature until the reaction was completed (
1H-NMR monitoring). After 4 h, perfluorinated imidazolium sulfonate bearing two epoxides
5 was isolated in 92% yield. Additionally, an excess of mCPBA (5 eq.) was also used as another oxidizing reagent to give 90% of
5 after 24 h at 40 °C in acetonitrile.
In this sequence, the perfluorinated imidazolium sulfonate
5 was synthesized in 54% overall yield from imidazole, 4-bromo-1-butene, 1H,1H,2H,2H-perfluooctyl iodide, and sodium 4-hydroxybenzenesulfonate. This procedure does not require any purification by silica gel column chromatography during the entire sequence. This efficient pathway is reproducible and can be performed on a large scale in the laboratory. The structure and the purity of this new ionic liquid was confirmed by nuclear magnetic resonance using
1H-NMR,
19F-NMR,
13C-NMR, DEPT, COSY, HSQC, HMBC and Infrared spectroscopy (see part I of the
Supplementary Materials). High-resolution mass spectrometry confirmed the two subparts of this salt with a cation [M]
+ at 485.0910 with the formula C
15H
14N
2OF
13 and an anion [M]
− at 243.0324 with the formula C
10H
11O
5S.
The last part of this work was devoted to the thermal properties of this perfluorinated imidazolium salt. The thermal stability was carried out by thermogravimetric analysis (TGA) using a Perkin Elmer Pyris 1 TGA working under nitrogen atmosphere with flow rate 20 mL·min
−1 and heating 20 K·min
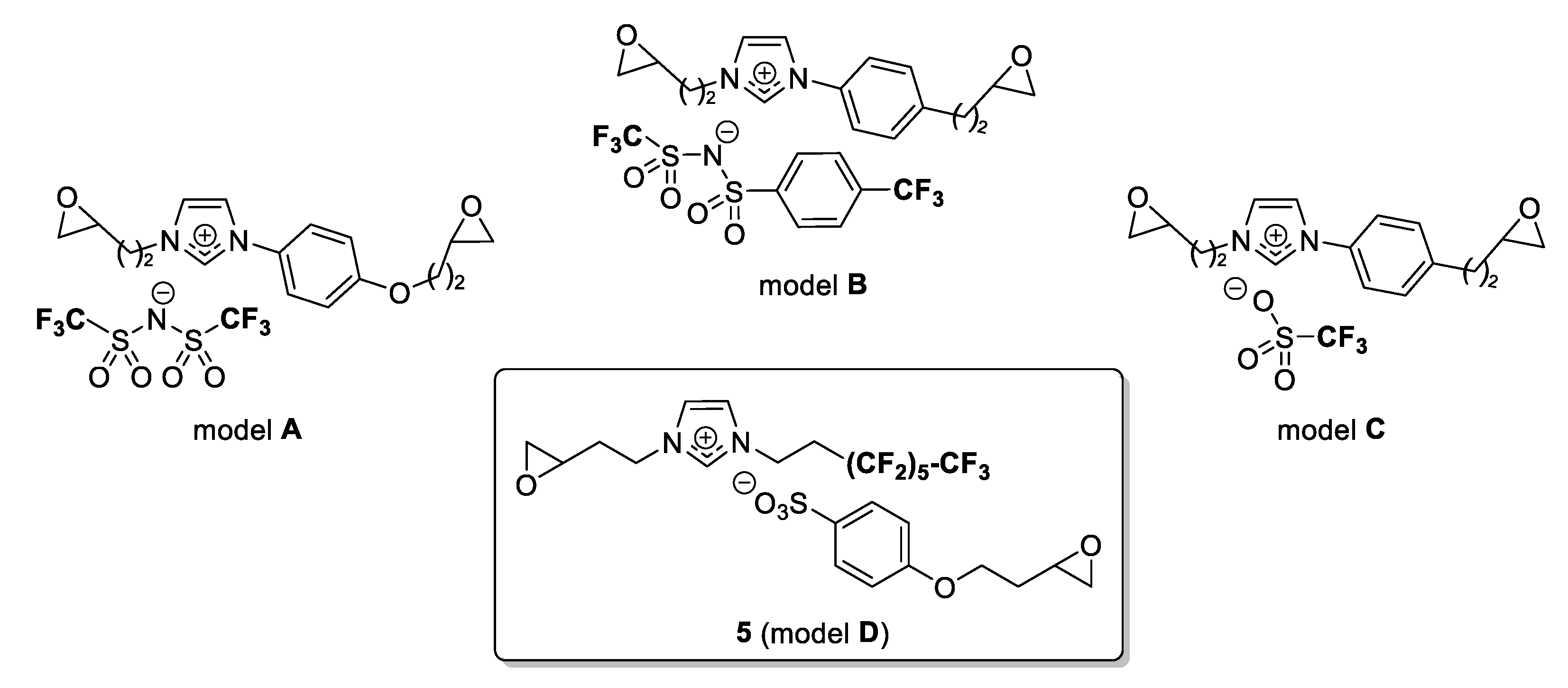
−1. The weight loss as a function of temperature was analyzed to determine the degradation temperature and the percentage of degradation of this salt. In previous studies, we reported the excellent thermal behavior for several diepoxy aryl-imidazolium triflimide salts (
Figure 1, model A) with a maximal degradation temperature above 390 °C. Homemade perfluorinated sulfonimides (
Figure 1, model B) afforded similar thermal stability. Very recently, new promising diepoxidized salts were described with sulfonate as a versatile counterion. In this unprecedented series, good thermal properties were observed around 375 °C to reach 411 °C with trifluoromethylsulfonate (
Figure 1, model C) as counter-anion [
13]. Here, by transferring the fluorinated chain to the imidazolium, the highly functionalized ionic liquid (
Figure 1, model D) showed a mass loss of only 10% at 245 °C to reach a T
max of 346 °C.
Finally, the thermal behavior of this salt was investigated by differential scanning calorimetry to determine the glass transition temperature, i.e., T
g. A glass transition temperature of −32 °C was obtained corresponding to the Tgs obtained in our previous studies [
9,
10,
11] opening some perspectives in the field of solid electrolytes where a lower Tg is required to reach the optimal ionic conductivity.
3. Materials and Methods
3.1. Chemistry
All reagents were purchased from Sigma Aldrich, Alfa Aesar, Acros Organic or TCI and were used as received: mCPBA (≤77% from Sigma Aldrich), 4-bromo-1-butene (97% from Alfa Aesar), anisole (>99% from Sigma Aldrich), potassium carbonate (99% from Acros Organic), imidazole (from TCI), perfluooctyle iodide (96% from Sigma Aldrich), sodium hydride (60% from Sigma Aldrich), Sodium 4-Hydroxybenzenesulfonate (<98% from TCI). DMDO was prepared according to the procedure described by D. F. Taber [
12]. Solvents were used in RPE grade without further purification. Anhydrous solvents were obtained from a PURESOLV SPS400 apparatus developed by Innovative Technology Inc.
1H-,
19F- and
13C-NMR spectra were recorded on a Bruker Avance III 500 MHz or Avance NEO 600 MHz spectrometer. Samples were dissolved in an appropriate deuterated solvent (CDCl
3, CD
3CN, D
2O and acetone-d
6). The chemical shifts (δ) are expressed in ppm relative to internal tetramethylsilane for
1H and
13C nuclei and coupling constants are indicated in Hz. Abbreviations for signal coupling are as follows: s = singlet; d = doublet; dd = doublet of doublets; t = triplet; q = quartet; m = multiplet. To assign the signals to the different proton and carbon atoms, additional 2D NMR experiments (COSY, HSQC, HMBC and DEPT) were performed. High-resolution mass spectra (HRMS) were performed on Acquity UPLC H-Class Xevo G2-XS QTof (WATERS) by electrospray ionization (ESI). Infrared (IR) spectra were recorded with a Perkin Elmer 16 PC FTIR ATR spectrometer, using the pure product (oil or solid). Thermographic analyses were recorded with Perkin Elmer Pyris 1 TGA, working under azote atmosphere with flow rate 20 mL/min and heating 20 °C/min. Thin layer chromatography (TLC) was run on pre-coated aluminum plates of silica gel 60 F-254 (Merck). Flash chromatography was performed on silica gel column (Merck silica gel, 40–63 mm).
3.2. Synthesis of 1-(3-buten-1-yl)imidazole 1
To a round-bottom flask purged with Ar, imidazole (5.00 g, 73.5 mmol, 1.0 eq) was added to a suspension of NaH (5.88 g, 147 mmol, 2.0 eq) in dry THF (100 mL) at 0 °C. The reaction was stirred for 30 min and we completed the mixture with 4-bromobutene (1.06 mL, 10.54 mmol, 2.0 eq) and potassium iodide (0.61 g, 3.60 mmol, 0.05 eq). After 18 h at 60 °C, the solvent was removed under reduced pressure. The residue was partitioned between CH2Cl2 and water and the organic layer was washed with NH4Cl saturated (2 × 10 mL). The combined organic extracts were dried over MgSO4, and concentrated under reduced pressure to afford product 1 as a clear yellow oil (6.45 g, 72%).
1H-NMR (600 MHz, CDCl3) δ 7.44 (s, 1H), 7.02 (s, 1H), 6.91 (s, 1H), 5.75–5.68 (m, 1H), 5.07–5.04 (m, 2H), 3.98 (t, J = 6.9 Hz, 2H), 2.50 (q, J = 6.9 Hz, 2H).
13C-NMR (151 MHz, CDCl3) δ 136.9, 133.5, 129.1, 118.7, 118.0, 46.3, 35.2.
IR (neat) cm−1 3379, 3108, 2979, 1641, 1507, 1438, 1359, 1283, 1228, 1108, 1077, 1036, 907, 814.
HRMS m/z (ESI): calcd. for C7H11N2 [MH]+: 123.0922, found: 123.0923.
3.3. Synthesis of 1-[1H,1H,2H,2H-perfluooctyl]-3-(3-buten-1-yl)imidazolium Iodide 2
To a solution of 1-(3-buten-1-yl)imidazole (0.24 g, 2 mmol, 1.0 eq) in CH3CN (10 mL) was added 1H,1H,2H,2H-perfluooctyl iodide (0.9 mL, 4 mmol, 2.0 eq). The mixture was refluxed at 80 °C and regularly monitored by 1H-NMR or TLC with a solution of dichloromethane/methanol (9/1). After cooling to room temperature, the solvent was removed under reduced pressure. The residue was partitioned between CH2Cl2 and water and the organic layer was washed with HCl (1M) (2 × 10 mL). The combined organic extracts were dried over MgSO4, and concentrated under reduced pressure to afford product 2 as a clear yellow oil (1.04 g, 89%). If necessary, the product can be purified by flash chromatography on silica gel with CH2Cl2/MeOH (9/1).
1H-NMR (600 MHz, CDCl3) δ 10.24 (s, 1H), 7.56 (s, 1H), 7.36 (s, 1H), 5.83–5.76 (m, 1H), 5.14–5.08 (m, 2H), 4.87 (t, J = 6.5 Hz, 2H), 4.30 (t, J = 6.7 Hz, 2H), 2.98–2.90 (m, 2H), 2.72–2.69 (m, 2H).
13C-NMR (151 MHz, CDCl3) δ 137.4, 131.9, 122.7, 122.1, 120.1, 118.1 (CF3), 116.1 (5 CF2), 49.7, 42.6, 34.2, 31.9.
19F-NMR (376 MHz, CDCl3) δ −80.8, −113.4, −121.8, −122.8, −123.2, −126.2.
IR (neat) cm−1 3054, 2882, 1772, 1563, 1422, 1265, 1240, 1145, 896, 735.
HRMS m/z (ESI): calcd. for C15H14F13N2 [M]+: 469.0949, found: 469.0953.
3.4. Synthesis of Sodium 4-(3-buten-1-yloxy)benzenesulfonate 3
To a solution of sodium 4-hydroxybenzenesulfonate (3.0 g, 12.9 mmol, 1 eq) and potassium carbonate (4.10 g, 117 mmol 2.0 eq) in methanol (30 mL) was added 4-bromobut-1-ene (4.06 mL, 40.1 mmol, 3.1 eq). The reaction mixture was stirred at 65 °C for 4 days. The solid residue was filtered and the solvent was concentrated under pressure. The product 3 was obtained as a white solid (2.92 g, 89%).
1H-NMR (500 MHz, D2O) δ 7.66 (d, J = 8.5 Hz, 2H), 6.99 (d, J = 8.5 Hz, 2H), 5.90–5.82 (m, 2H), 5.14–5.04 (m, 1H), 4.11 (t, J = 6.4 Hz, 2H), 2.49–2.45 (m, 2H).
13C-NMR (126 MHz, D2O) δ 160.5, 135.2, 127.5 (×2), 117.2, 115.6, 115.0 (×2), 67.9, 32.8.
IR (neat) cm−1 3432, 3059, 2970, 1738, 1643, 1638, 1499, 1473, 1231, 1180, 1139, 1065, 1031, 1007, 900.
Mp: >300 °C
HRMS m/z (ESI): calcd. for C10H11O4S [M]−: 227.0378, found: 227.0370.
3.5. Synthesis of 1-[1H,1H,2H,2H-perfluooctyl]-3-(3-buten-1-yl)imidazolium 4-(3-buten-1-yloxy)benzenesulfonate 4
To a stirred solution of sodium sulfonate 3 (40 mg, 0.16 mmol, 1.0 eq) in biphasic solution CH2Cl2/H2O (1:1, v/v) (8 mL) was added imidazolium iodide 2 (100 mg, 0.16 mmol, 1.0 eq). The mixture was stirred at room temperature for 24 h. The organic compounds were extracted with dichloromethane, dried with MgSO4 and concentrated under reduced pressure. After NMR analysis, an excess of sodium sulfonate (1.0 eq) was used to complete the reaction. This mixture was stirred at room temperature for another 24 h. The product 4 was extracted with dichloromethane, dried with MgSO4 and then concentrated under reduced pressure to afford a yellow clear oil (101 mg, 91%).
1H-NMR (600 MHz, CDCl3) δ 9.82 (s, 1H), 7.77 (d, J = 8.8 Hz, 2H), 7.58 (s, 1H), 7.37 (s, 1H), 6.83 (d, J = 8.8 Hz, 2H), 5.90–5.84 (m, 1H), 5.70–5.63 (m, 1H), 5.16–5.08 (m, 2H), 5.02–4.95 (m, 2H), 4.70 (t, J = 6.5 Hz, 2H), 4.28 (t, J = 6.8 Hz, 2H), 3.98 (t, J = 6.7 Hz, 2H), 2.81–2.73 (m, 2H), 2.56–2.50 (m, 4H).
13C-NMR (151 MHz, CDCl3) δ 159.9, 138.7, 137.8 (×2), 134.2, 132.2 (×2), 127.4, 122.8, 122.2, 119.5, 118.1 (CF3), 117.1, 110.8 (5 CF2), 113.9, 67.3, 49.2, 42.2, 34.2, 33.5, 26.9.
19F-NMR (376 MHz, CDCl3) δ −85.8, −118.46, −126.8, −127.9, −128.2, −131.2.
IR (neat) cm−1 3050, 2772, 1780, 1610, 1423, 1265, 1240, 1188, 1121, 1078, 1004, 835, 735.
HRMS m/z (ESI): calcd. for C15H14F13N2 [M]+: 469.0949, found: 469.0958, calcd. for C10H11O4S [M]−: 227.0378, found: 227.0378.
3.6. Synthesis of 1-[1H,1H,2H,2H-perfluooctyl]-3-[2-(Oxiran-2-yl)ethyl]imidazolium 4-[(2-oxiran-2-yl)ethoxy]benzenesulfonate 5
To a solution of compound 4 (50 mg, 0.071 mmol, 1.0 eq) in acetone (0.50 mL) freshly prepared DMDO (0.044 mol/L) (4.57 mL, 0.20 mmol, 2.8 eq) was added. The reaction mixture was stirred at room temperature for 4 h. Two drops of DMS were added to neutralize the excess of DMDO. The solvent was evaporated under reduced pressure and the product 5 was obtained as a red clear oil (48 mg, 92%).
1H-NMR (500 MHz, CD3CN) δ 8.73 (s, 1H), 7.56 (d, J = 9.2 Hz, 2H), 7.41 (s, 1H), 7.37 (s, 1H), 6.77 (d, J = 9.1 Hz, 2H), 4.45 (t, J = 7.2 Hz, 2H), 4.25–4.22 (m, 2H), 4.05–4.03 (m, 2H), 2.99–2.96 (m, 1H), 2.88–2.85 (m, 1H), 2.81–2.72 (m, 2H), 2.66–2.65 (dd, J = 4.7 and 4.0 Hz, 1H), 2.6–2.61 (dd, J = 4.7 and 4.0 Hz, 1H), 2.44–2.43 (dd, J = 5.0 and 2.7 Hz, 1H), 2.36–2.35 (dd, J = 5.0 and 2.7 Hz, 1H), 2.14–2.09 (m, 1H), 1.94–1.89 (m, 1H), 1.84–1.79 (m, 2H).
13C-NMR (126 MHz, CD3CN) δ 159.7, 141.4, 137.4, 127.8 (×2), 123.4, 123.2, 123.2 (CF3), 114.1 (×2), 111.2 (5 CF2), 65.5, 49.8, 49.3, 47.6, 46.9, 46.4, 42.2, 32.9 (×2), 32.7.
19F-NMR (376 MHz, CD3CN) δ −81.5, −114.4, −122.3, −123.3, −124.1, −126.6.
IR (neat) cm−1 3100, 2769, 1598; 1188, 1144, 1121, 1078, 1004, 961, 835, 809, 746, 699.
HRMS m/z (ESI): calcd. for C15H14N2OF13 [M]+: 485.0899, found: 485.0910, calcd. for C10H11O5S [M]−: 243.0327, found: 243.0324.




{kind=link}
{kind=link}