
2,6-exo-8,10-exo-4-Butyl-9-oxa-4-azatetracyclo[5.3.1.02,6.08,10]undecane-3,5-dione



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
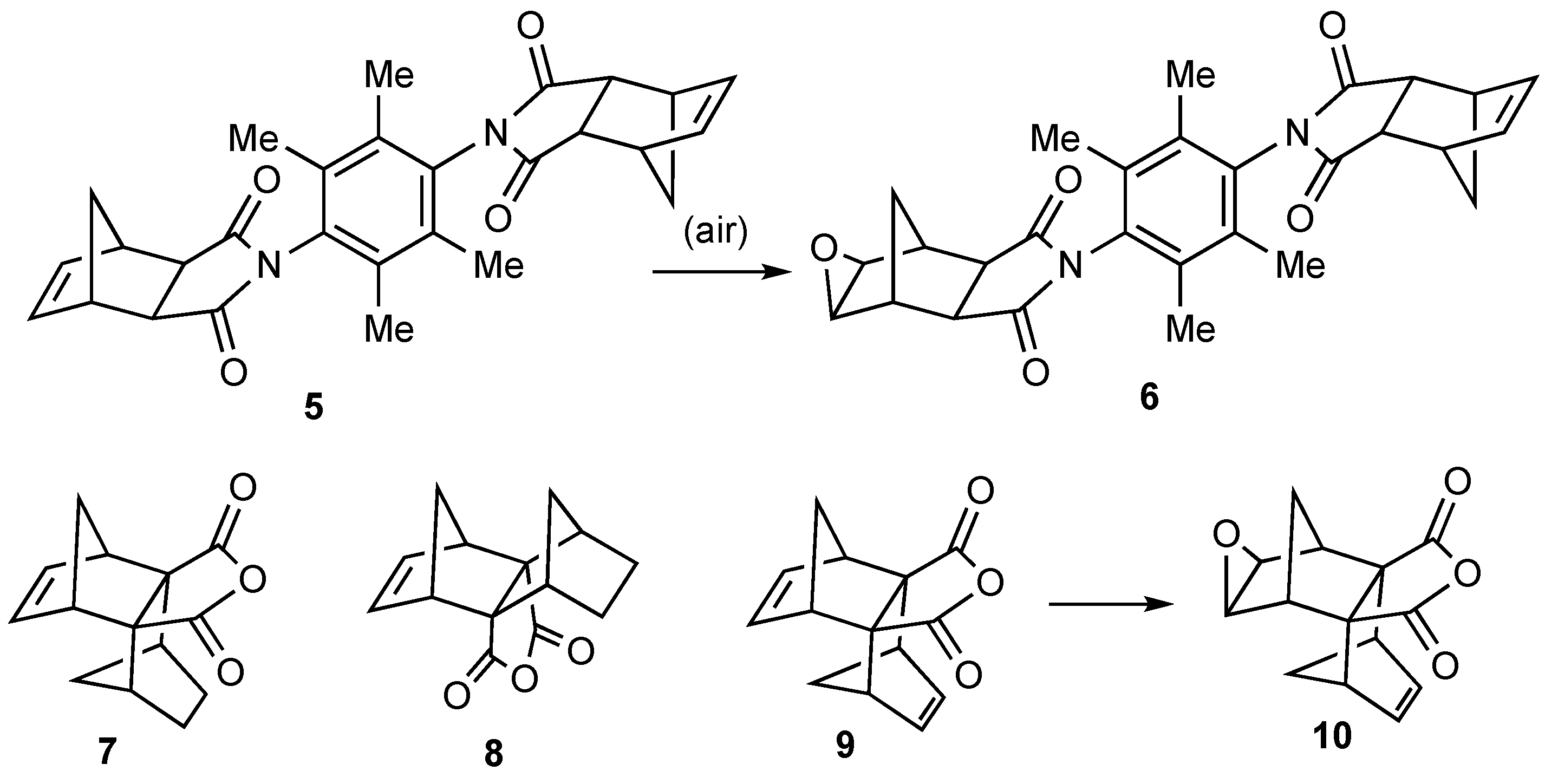
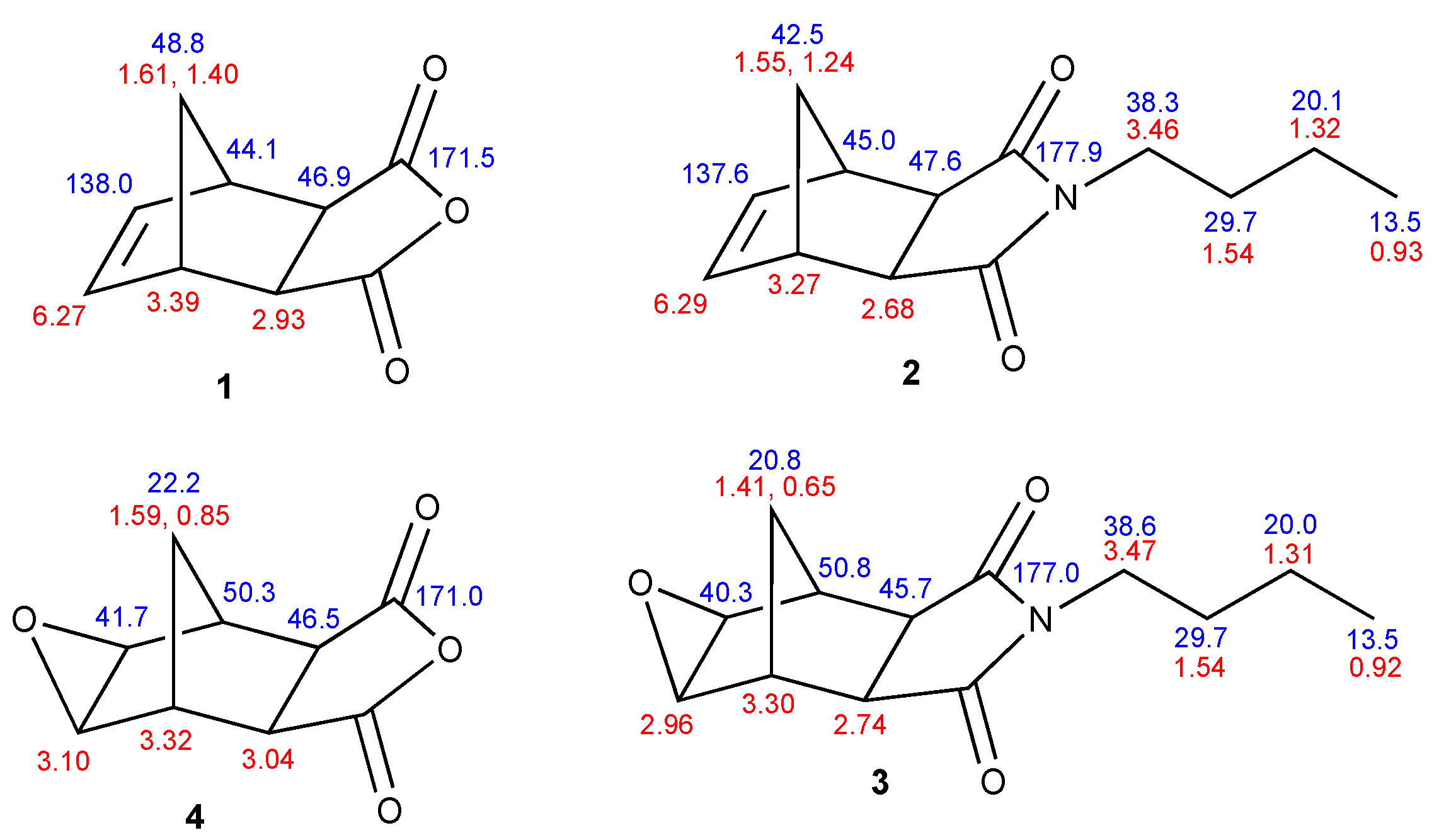
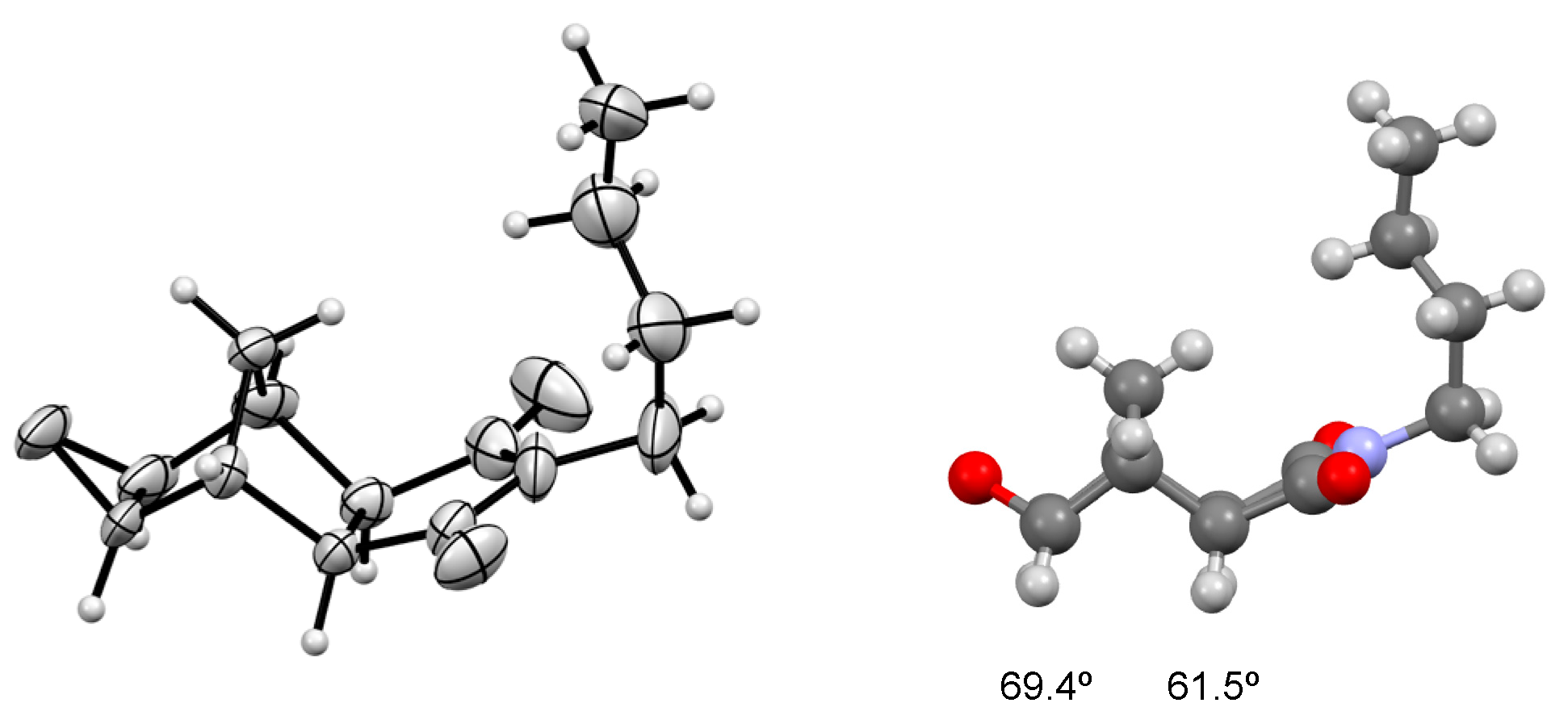
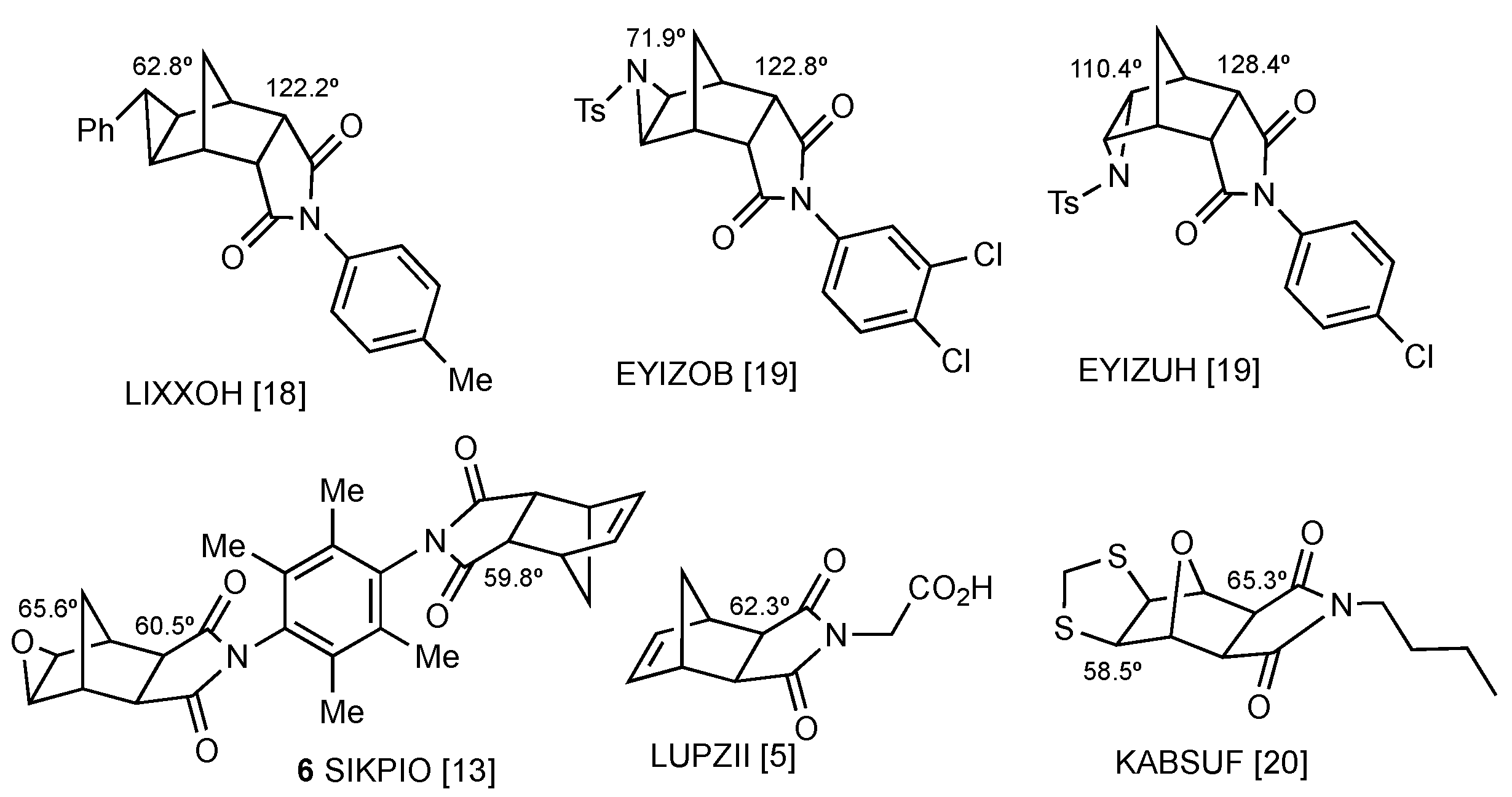
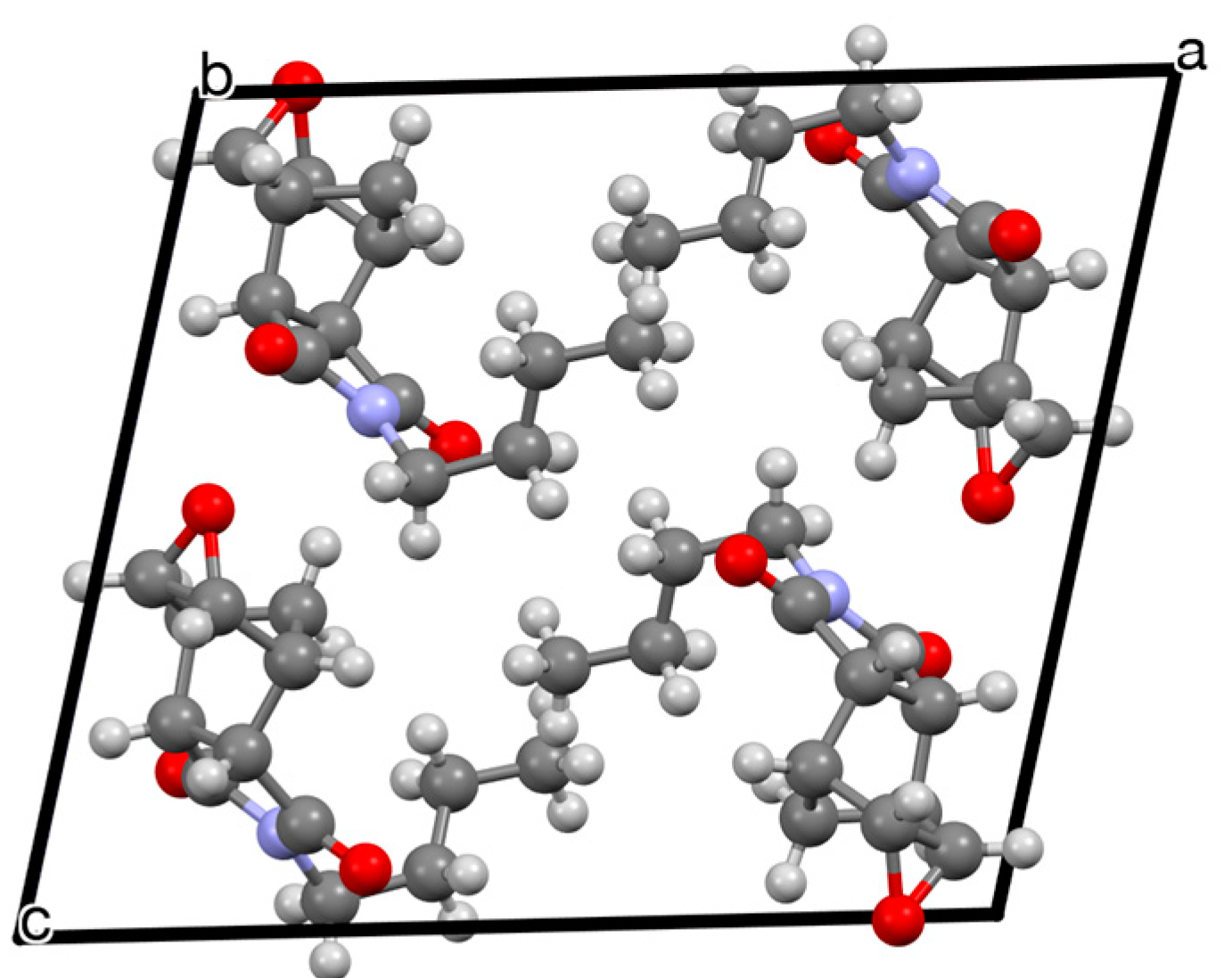
2. Results
3. Experimental
3.1. 2,6-exo-4-Butyl-4-azatricyclo[5.2.1.02,6]dec-8-ene-3,5-dione (2)
3.2. 2,6-exo-8,10-exo-4-Butyl-9-oxa-4-azatetracyclo[5.3.1.02,6.08,10]undecane-3,5-dione (3)
3.3. 2,6-exo-8,10-exo-4,9-dioxatetracyclo[5.3.1.02,6.08,10]undecane-3,5-dione (4)
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Luzzio, F.A. (Ed.) Imides: Medicinal, Agricultural, Synthetic Applications and Natural Products Chemistry; Elsevier: Oxford, UK, 2019; 456p. [Google Scholar]
- Khosravi, E.; Iqbal, F.; Musa, O.M. Thermosetting ROMP materials with thermally degradable linkages. Polymer 2011, 52, 243–249. [Google Scholar] [CrossRef]
- Liu, M.; van Hensbergen, J.; Burford, R.P.; Lowe, A.B. Thiol-Michael coupling chemistry: Facile access to a library of functional exo-7-oxanorbornenes and their ring-opening metathesis (co)polymerization. Polym. Chem. 2012, 3, 1647–1658. [Google Scholar] [CrossRef]
- Chang, A.B.; Lin, T.-P.; Thompson, N.B.; Luo, S.-X.; Liberman-Martin, A.L.; Chen, H.-Y.; Lee, B.; Grubbs, R.H. Design, synthesis and self-assembly of polymers with tailored graft distributions. J. Am. Chem. Soc. 2017, 139, 17683–17693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birchall, L.T.; Shehata, S.; McCarthy, S.; Shepherd, H.J.; Clark, E.R.; Serpell, C.J.; Baigini, S.C.G. Supramolecular behaviour and fluorescence of rhodamine-functionalised ROMP polymers. Polym. Chem. 2020, 11, 5279–5285. [Google Scholar] [CrossRef]
- Scappaticci, K.A. Treatment of Depression. US Patent 5011841, 30 April 1991. [Google Scholar]
- Aitken, R.A.; Carcas, K.; Hill, L.; Massil, T.; Raut, S.V. Cycloaddition of Bun3P•CS2: Direct One-pot Conversion of Strained Double Bonds to 2-Alkylidene-1,3-dithiolanes. Tetrahedron 1997, 53, 2261–2270. [Google Scholar] [CrossRef]
- Aitken, R.A.; Hill, L.; Lightfoot, P. Direct One Pot Construction of Norbornane-fused Dihydrotetrathiafulvalenes. Tetrahedron Lett. 1997, 38, 7927–7930. [Google Scholar] [CrossRef]
- Craig, D. The rearrangement of endo-3,6-methylene-1,2,3,6-tetrahydro-cis-phthalic anhydride. J. Am. Chem. Soc. 1951, 73, 4889–4892. [Google Scholar] [CrossRef]
- Murphy, R.B.; Pham, D.-T.; White, J.M.; Lincoln, S.F.; Johnston, M.R. Molecular tweezers with a rotationally restricted linker and freely rotating porphyrin moieties. Org. Biomol. Chem. 2018, 16, 6206–6223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartlett, P.D.; Combs, G., Jr.; Le, A.-X.T.; Watson, W.H.; Galloy, J.; Kimura, M. Reactivities of norbornene-type double bonds in propellane derivatives and their precursors; furan as a diene. J. Am. Chem. Soc. 1982, 104, 3131–3138. [Google Scholar] [CrossRef]
- Petrova, T.; Tarabara, I.; Palchikov, V.; Kasyan, L.; Kosenkov, D.; Okovytyy, S.; Gorb, L.; Shishkina, S.; Shishkin, O.; Leszczynski, J. Ethanolysis of N-substituted norbornane epoxyimides: Discovery of diverse pathways depending on substitu-ent’s character. Org. Biomol. Chem. 2010, 8, 2142–2157. [Google Scholar] [CrossRef]
- Aitken, R.A.; Gosney, I.; Farries, H.; Palmer, M.H.; Simpson, I.; Cadogan, J.I.G.; Tinley, E.J. Chemical repercussions of orbital interactions through bond and through space. The reactivity of the double bond in unsaturated cyclic sulphones towards aziridine formation and epoxidation. Tetrahedron. 1984, 40, 2487–2503. [Google Scholar] [CrossRef]
- Berson, J.A.; Suzuki, S. The hydration of exo-cis-3,5-endomethylene Δ4-tetrahydrophthalic anhydride. J. Am. Chem. Soc. 1958, 80, 4341–4345. [Google Scholar] [CrossRef]
- Kas’yan, L.I.; Krishchik, O.V.; Tarabara, I.N.; Kas’yan, A.O.; Pal’chikov, V.A. Lactonization of epoxyendic anhydride in re-actions with amines. Russ. J. Org. Chem. 2006, 42, 501–508. [Google Scholar] [CrossRef]
- Meijer, A.; Otoo, S.; Engberts, J.B.F.N. Effects of the hydrophobicity of the reactants on Diels-Alder reactions in water. J. Org. Chem. 1998, 63, 8989–8994. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.; Kim, M.; Lee, J.; Do, Y.; Chang, S. Trimanganese complexes bearing bidentate nitrogen ligands as a highly efficient catalyst precursor in the epoxidation of alkenes. J. Org. Chem. 2006, 71, 6721–6727. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Zhang, S.-Q.; Shi, B.-F.; Bao, W. Palladium(0)-catalyzed cyclopropanation of benzyl bromides via C(sp3)H bond acti-vation. Chem. Commun. 2014, 50, 3692–3694. [Google Scholar] [CrossRef] [PubMed]
- Bortoluzzi, A.J.; Andrade, E.S.; Nunes, R.J. 4-(3,4-Dichlorophenyl)-9-(4-methylphenylsulfonyl)-4,9-diazatetracyclo[5.3.1.02,6.08,10]undecane-3,5-dione and 4-(4-chlorophenyl)-9-(4-methylphenylsulfonyl)-4,9-diazatetracyclo[5.3.1.02,6.08,10]undecane-3,5-dione. Acta Crystallogr. Sect. C 2004, 60, o614–o616. [Google Scholar] [CrossRef] [PubMed]
- Aitken, R.A.; Fotherby, F.M.; Slawin, A.M.Z. 2,6-exo-8,12-exo-10-Butyl-13-oxa-3,5-dithia-10-azatetracyclo[5.5.1.02,6.08,12]tridecane-9,11-dione. Molbank 2020, 2020, M1123. [Google Scholar] [CrossRef] [Green Version]
- Aitken, R.A.; Sonecha, D.K. The solid-state structures of cyclic NH carboximides. Crystals 2020, 10, 606. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELXL. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aitken, R.A.; Fotherby, F.M.; Slawin, A.M.Z. 2,6-exo-8,10-exo-4-Butyl-9-oxa-4-azatetracyclo[5.3.1.02,6.08,10]undecane-3,5-dione. Molbank 2022, 2022, M1320. https://doi.org/10.3390/M1320
Aitken RA, Fotherby FM, Slawin AMZ. 2,6-exo-8,10-exo-4-Butyl-9-oxa-4-azatetracyclo[5.3.1.02,6.08,10]undecane-3,5-dione. Molbank. 2022; 2022(1):M1320. https://doi.org/10.3390/M1320
Chicago/Turabian StyleAitken, R. Alan, Fiona M. Fotherby, and Alexandra M. Z. Slawin. 2022. "2,6-exo-8,10-exo-4-Butyl-9-oxa-4-azatetracyclo[5.3.1.02,6.08,10]undecane-3,5-dione" Molbank 2022, no. 1: M1320. https://doi.org/10.3390/M1320
APA StyleAitken, R. A., Fotherby, F. M., & Slawin, A. M. Z. (2022). 2,6-exo-8,10-exo-4-Butyl-9-oxa-4-azatetracyclo[5.3.1.02,6.08,10]undecane-3,5-dione. Molbank, 2022(1), M1320. https://doi.org/10.3390/M1320