
2-Chloro-4,6-bis{(E)-3-methoxy-4-[(4-methoxybenzyl)oxy]styryl}pyrimidine: Synthesis, Spectroscopic and Computational Evaluation

 ,
,  ,
, 

Abstract
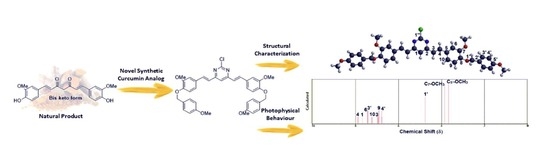
:
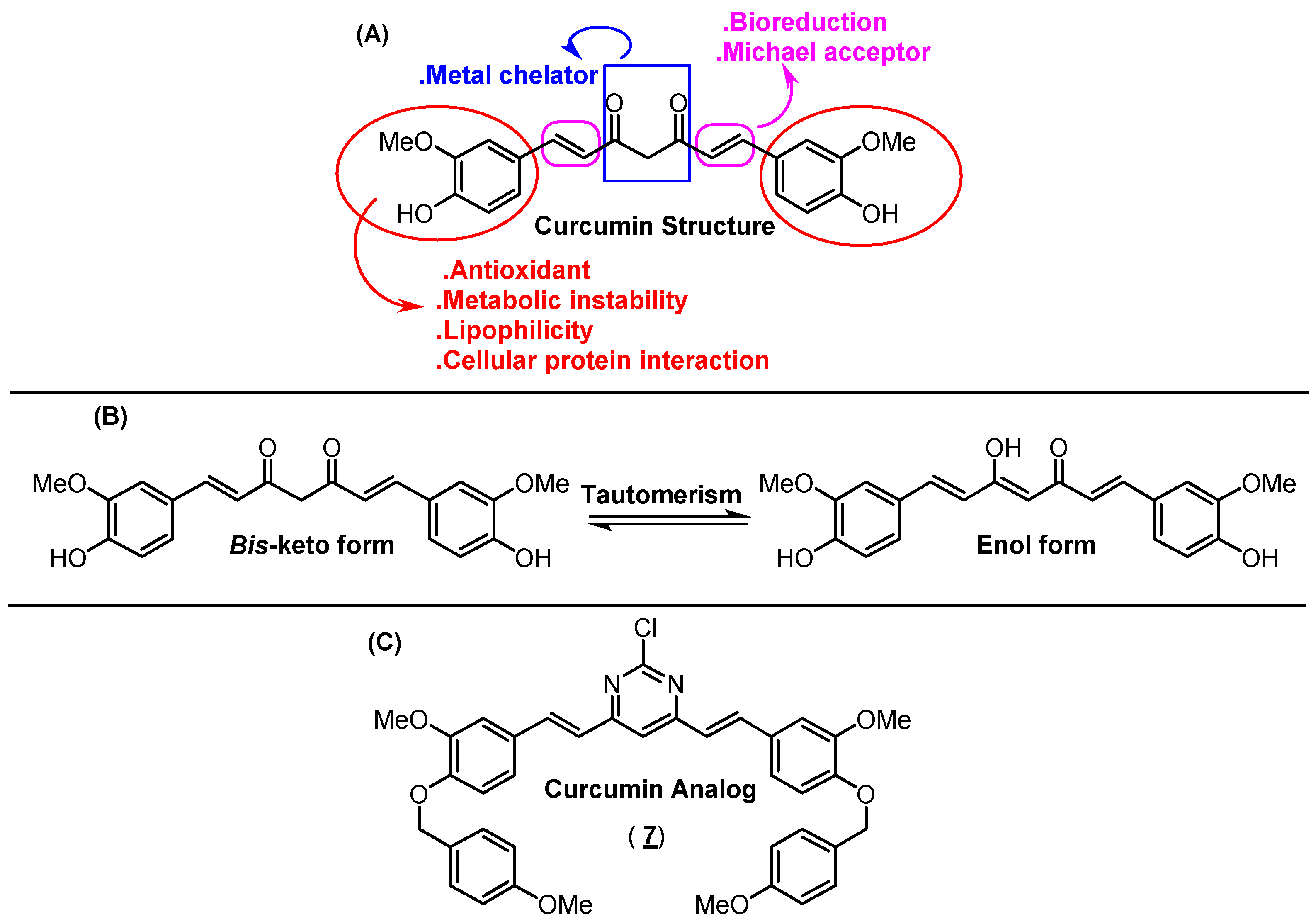
1. Introduction
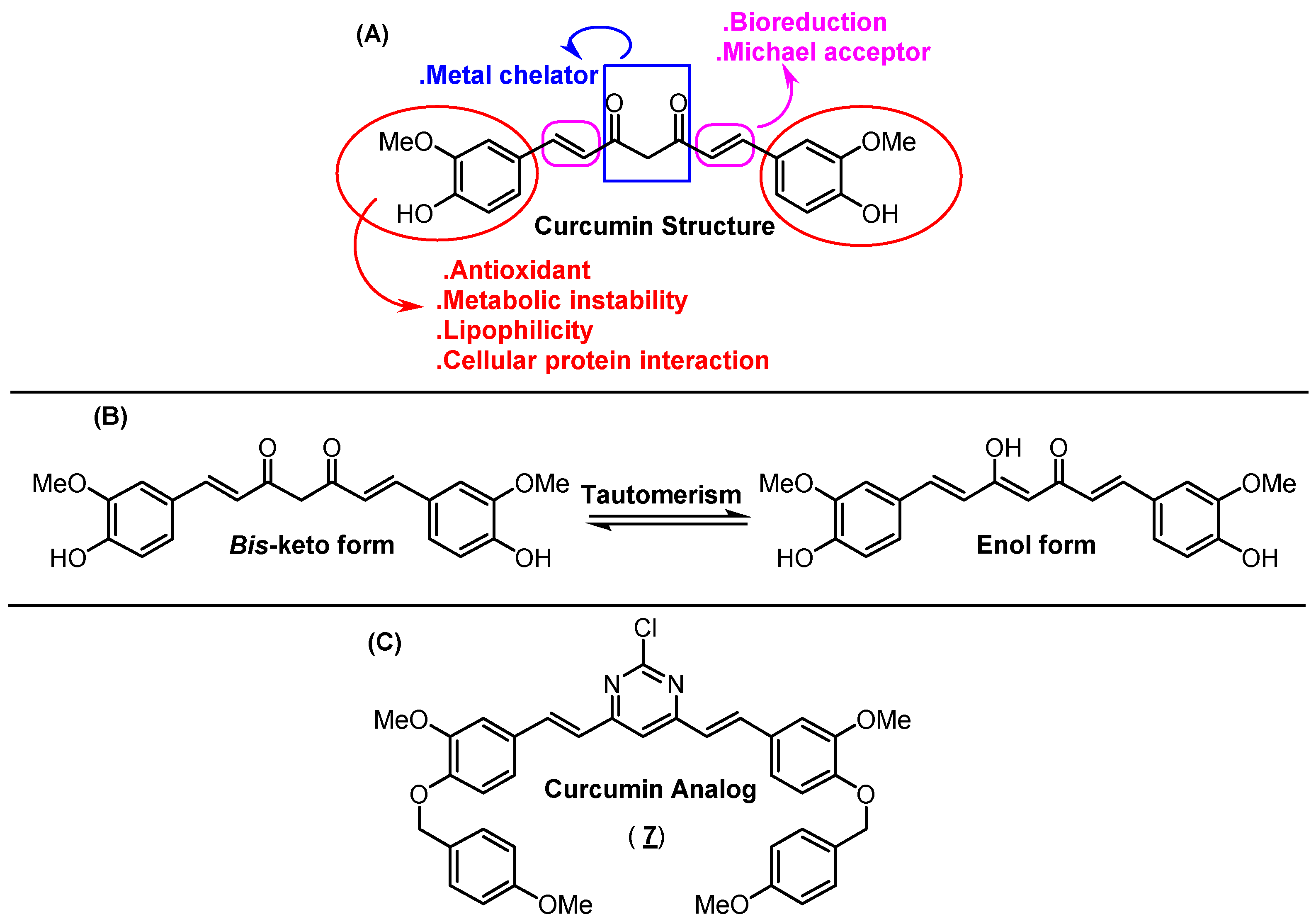
2. Results and Discussion
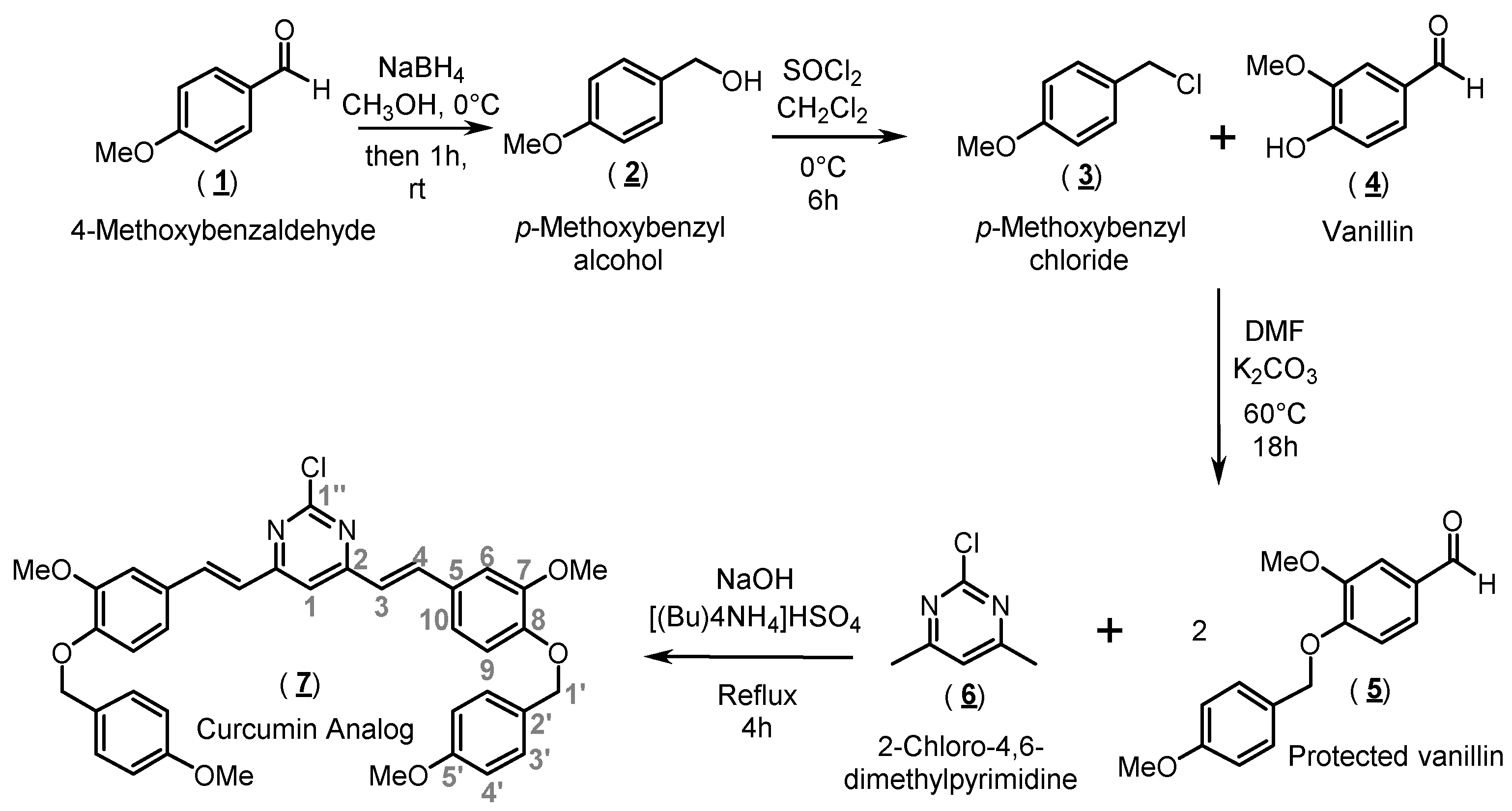
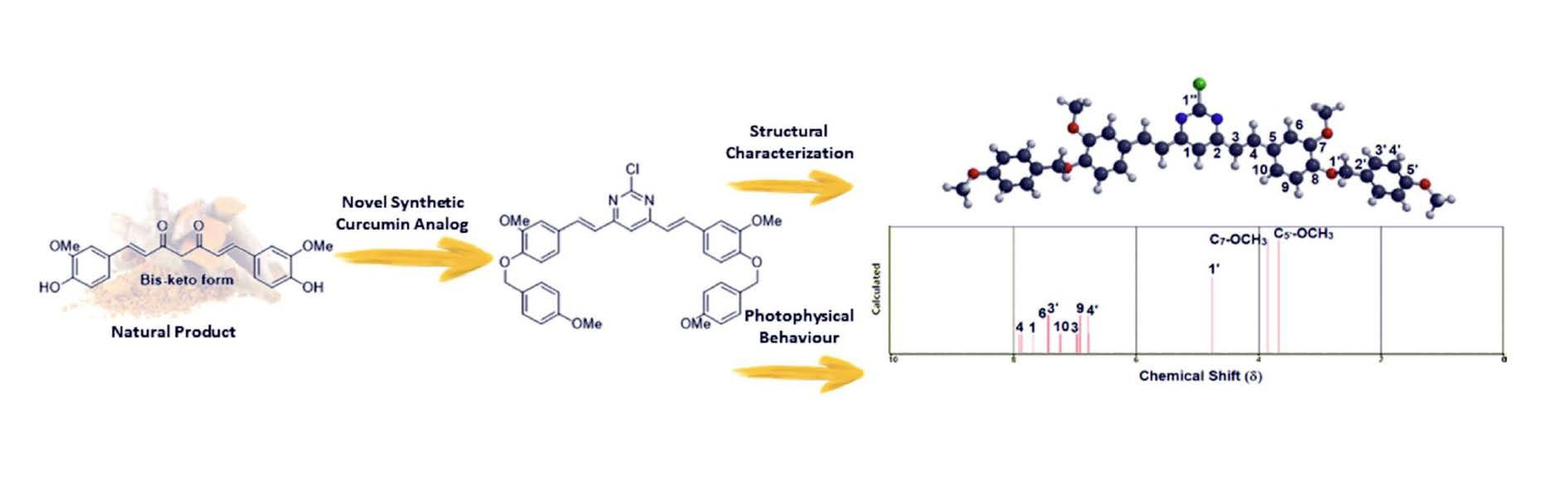
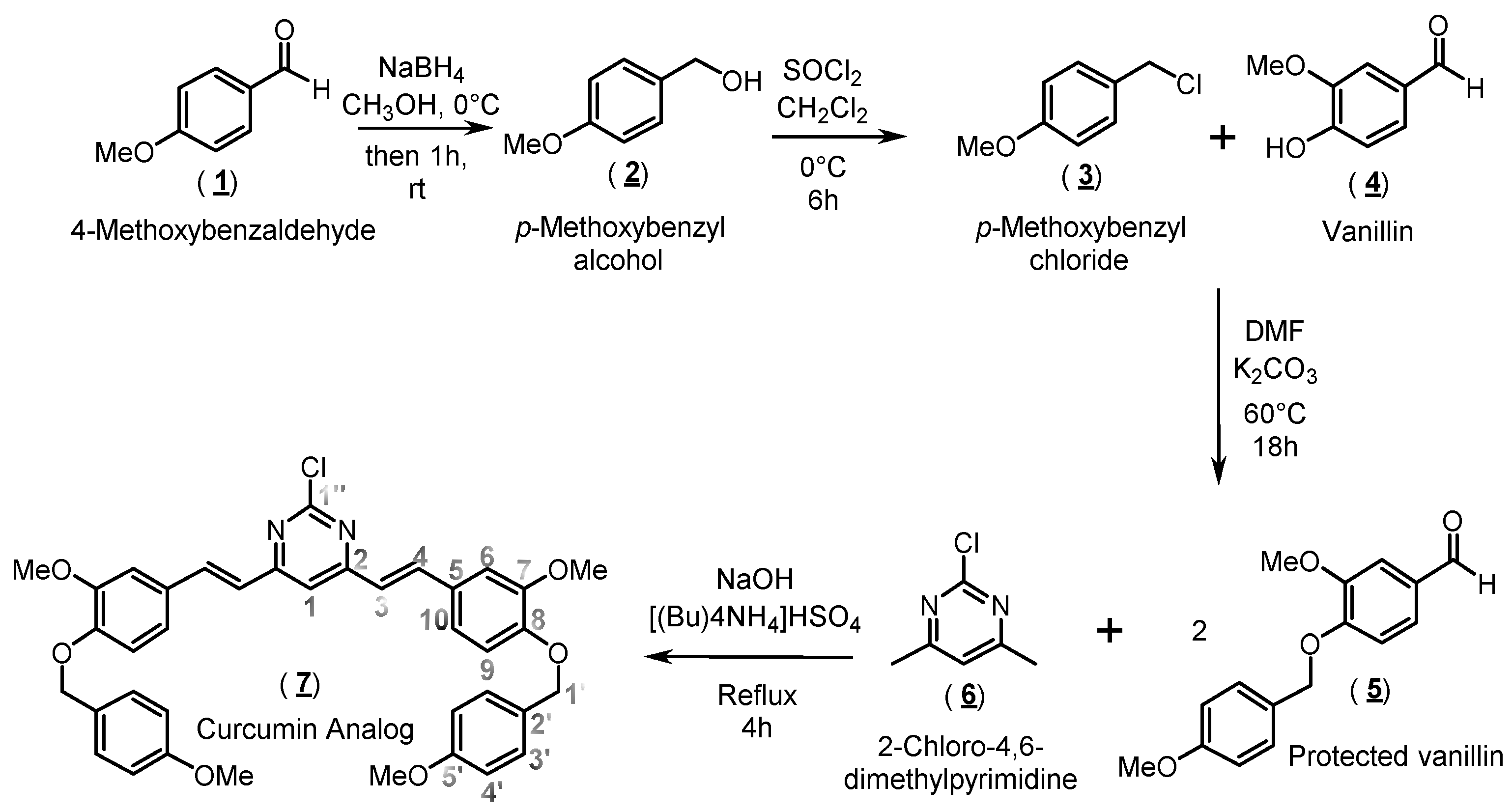
2.1. Organic Synthesis and Structure Determination
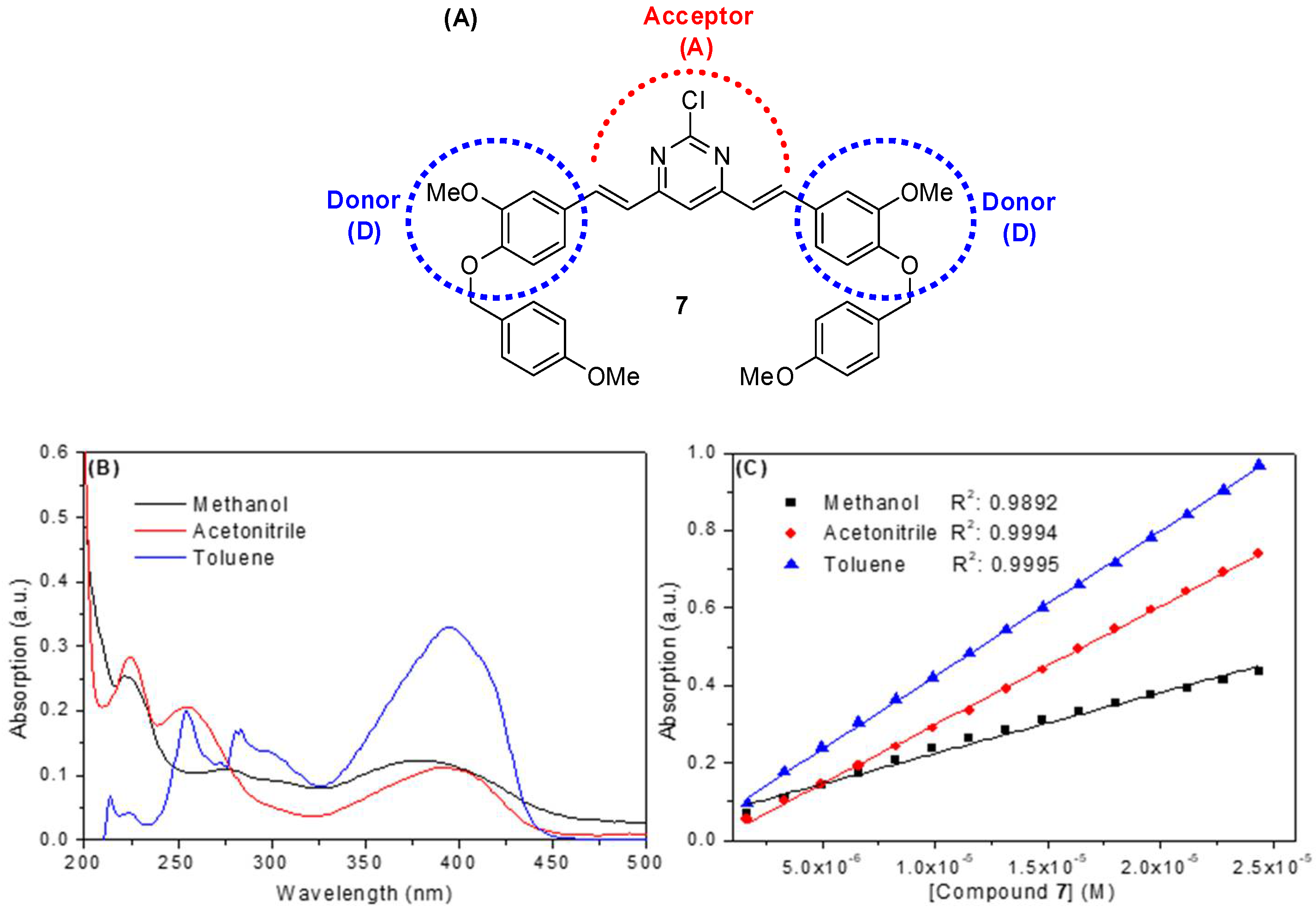
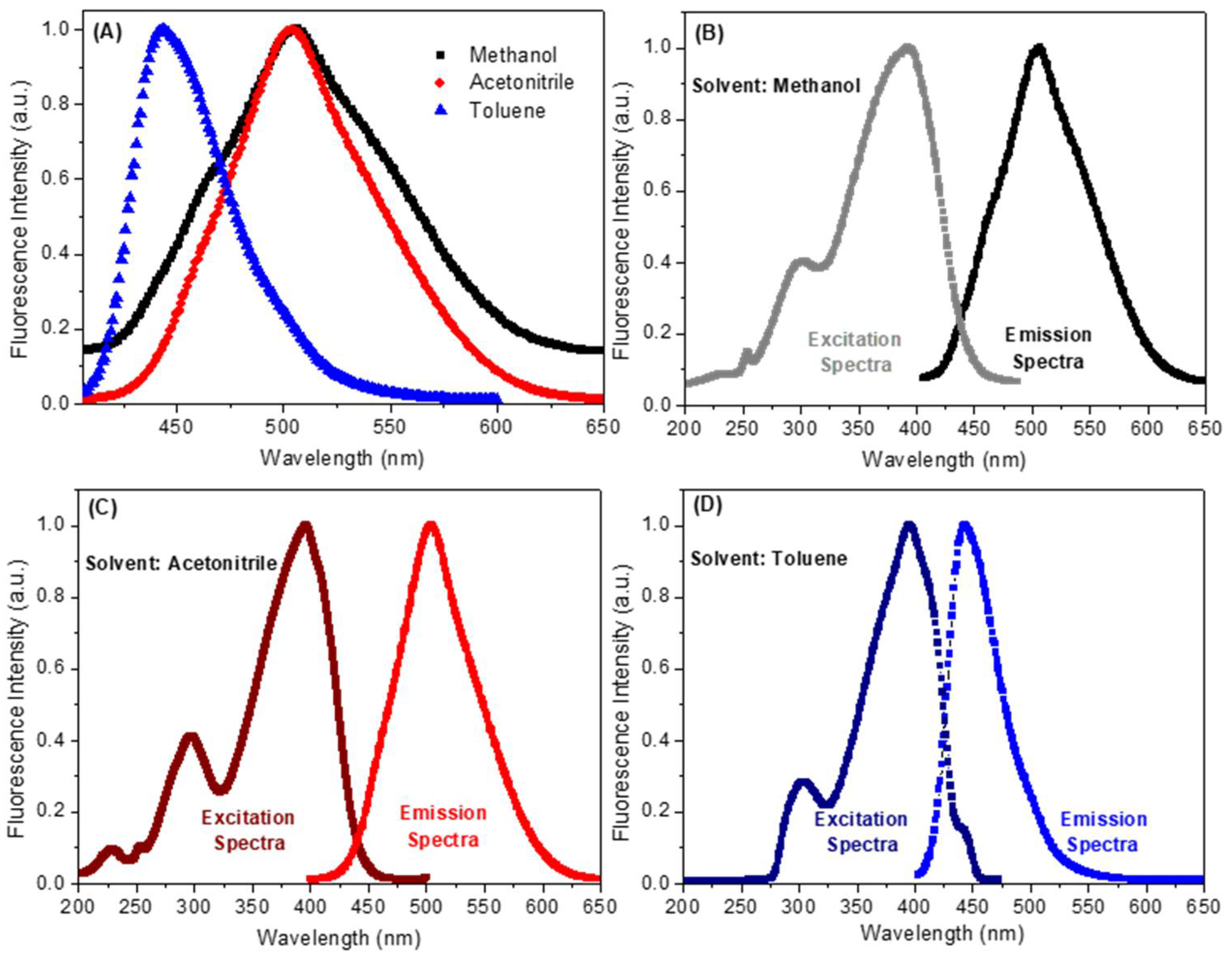
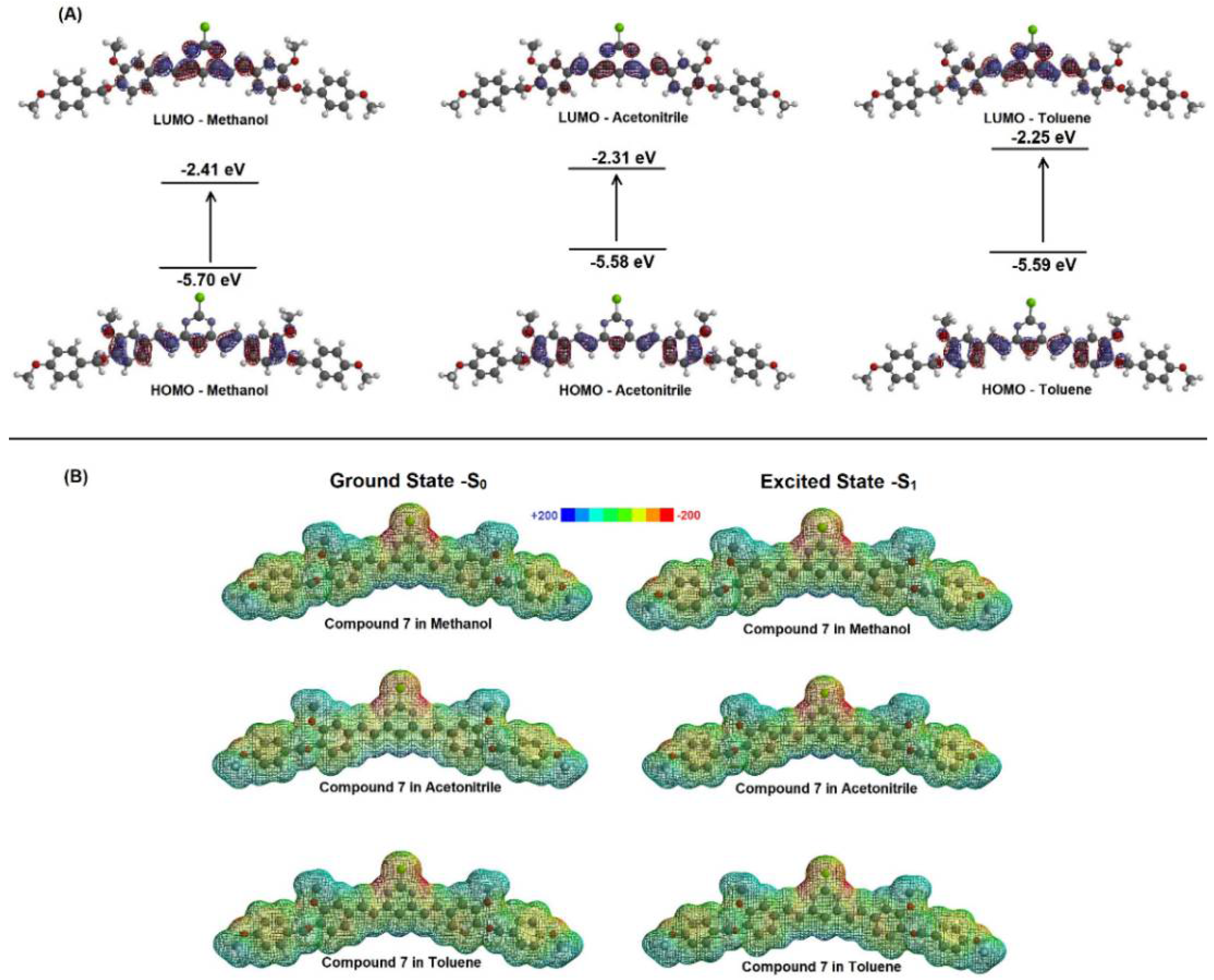
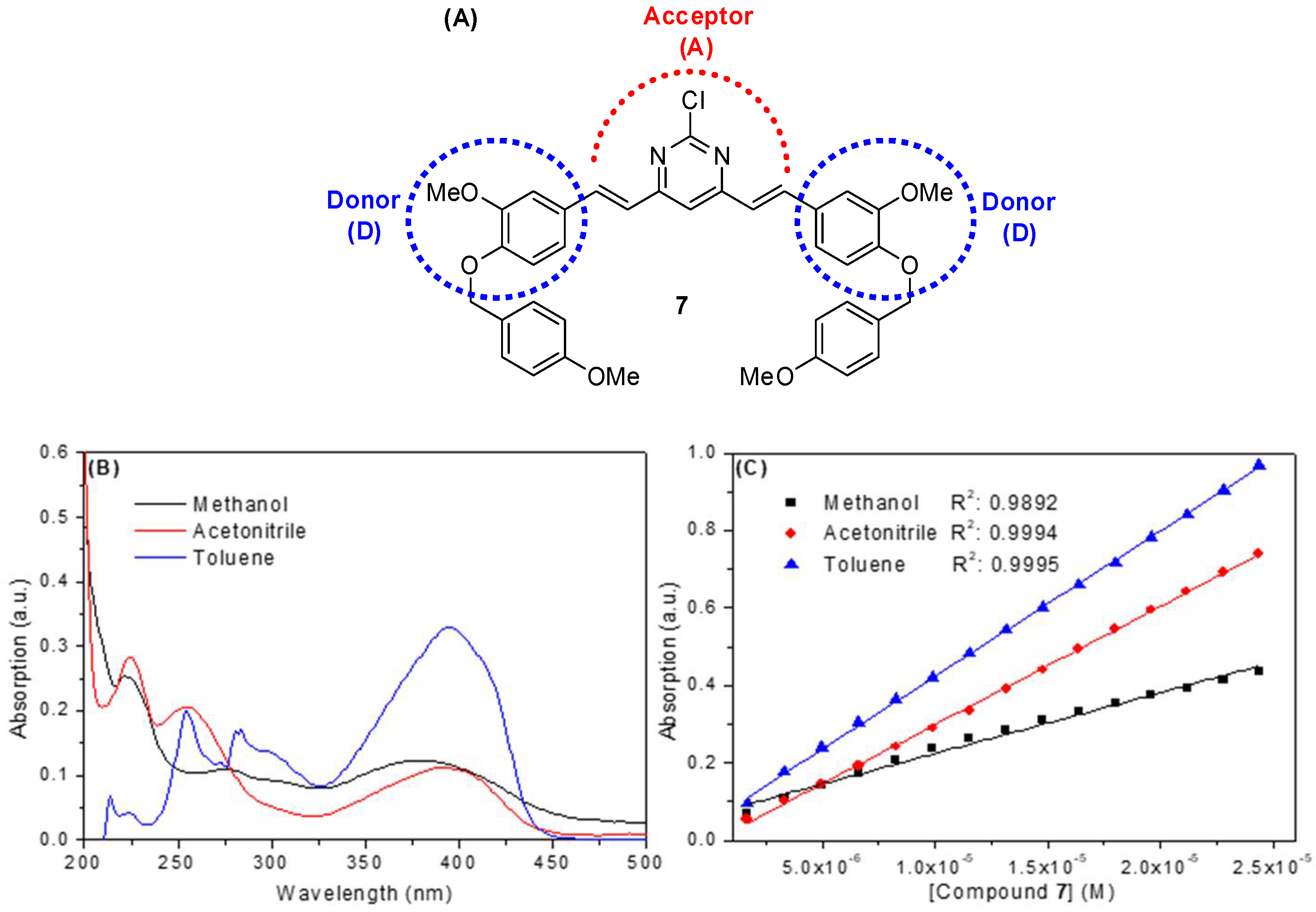
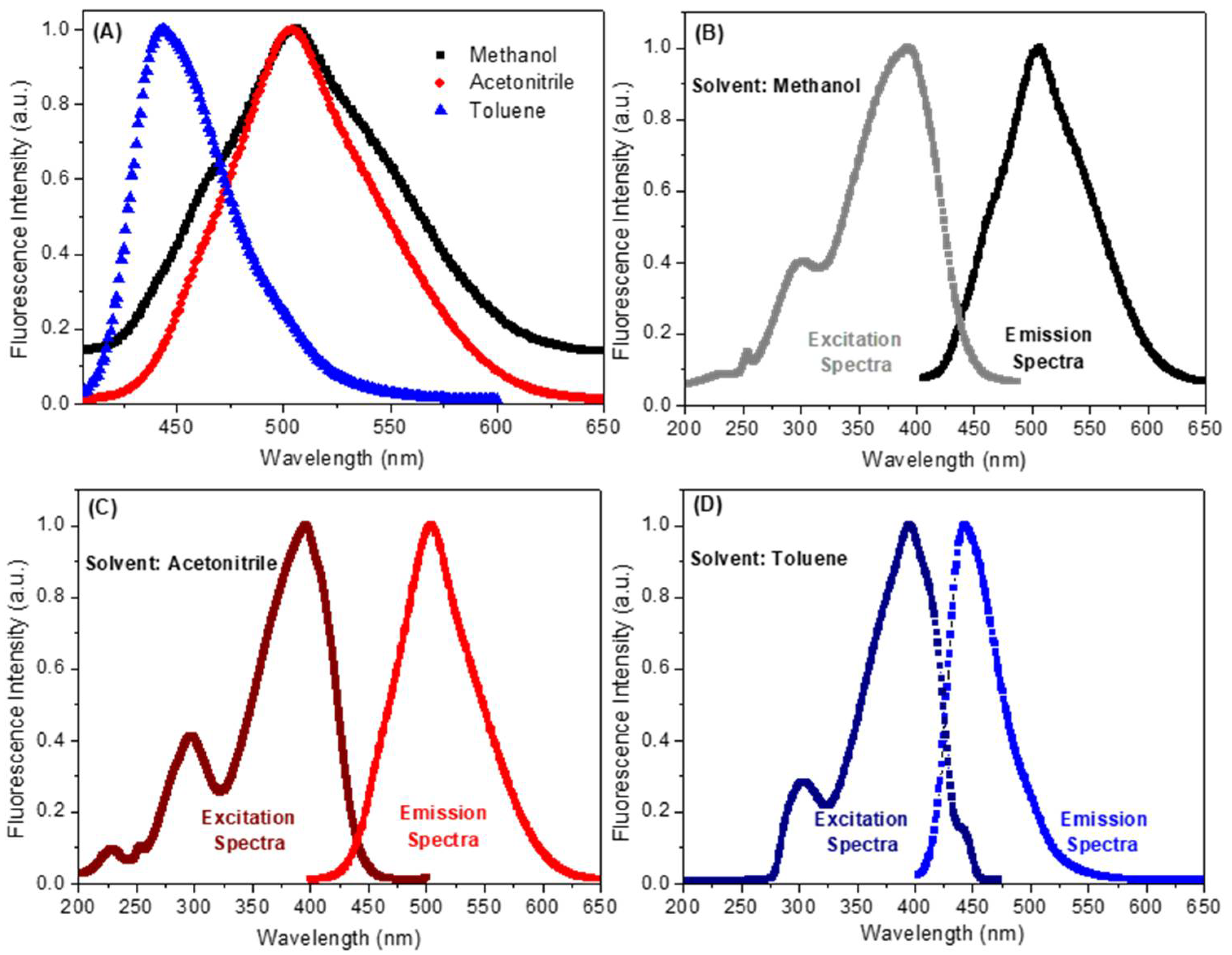
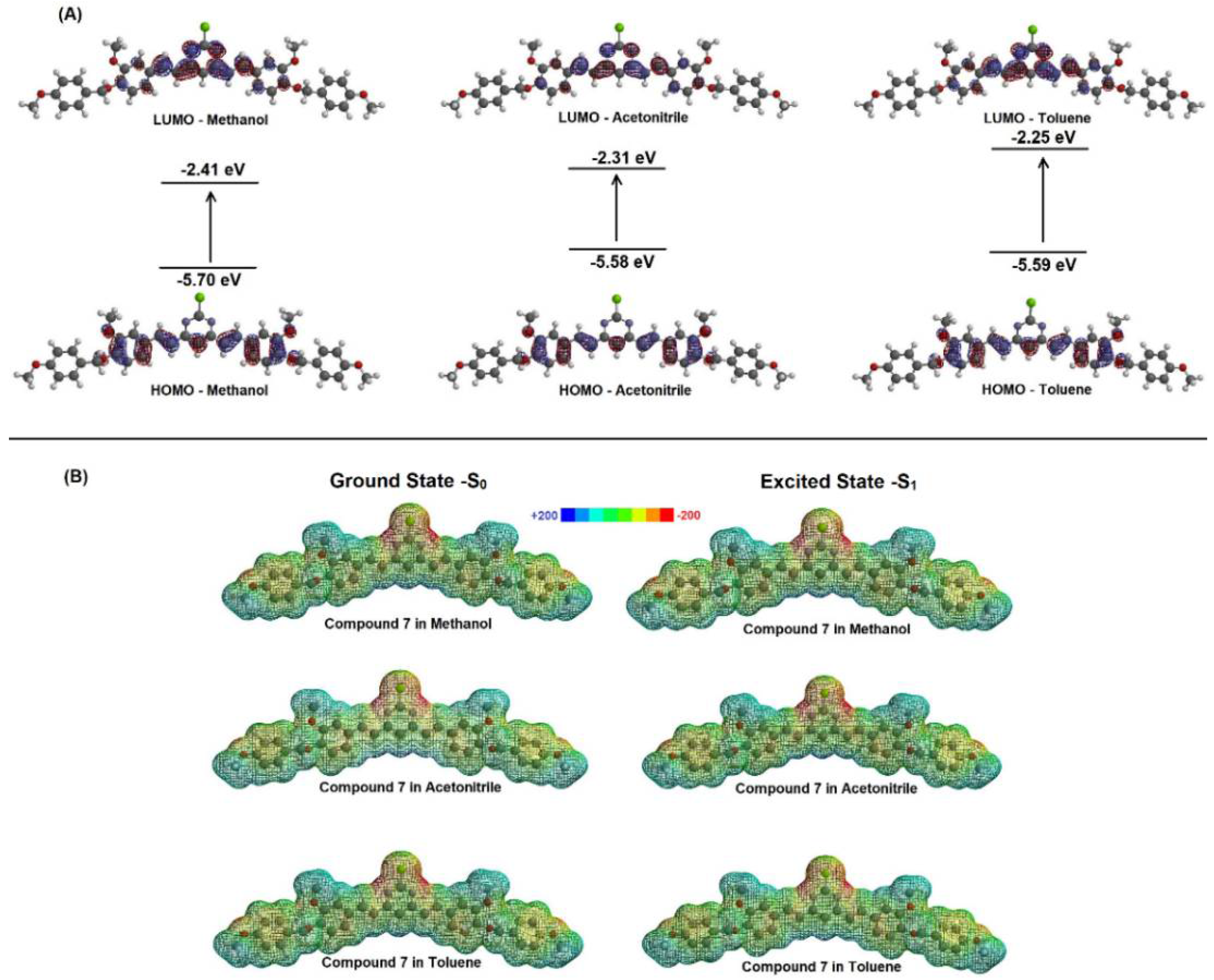
2.2. Spectroscopic Study: UV-Vis Absorption and Steady-State Fluorescence Emission
3. Materials and Methods
3.1. Synthesis of 2-Chloro-4,6-bis{(E)-3-methoxy-4-[(4-methoxybenzyl)oxy]-styryl}pyrimidine (Compound 7)
3.2. Spectroscopic Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sueth-Santiago, V.; Mendes-Silva, G.P.; Decoté-Ricardo, D.; de Lima, M.E.F. Curcumin, the golden powder from turmeric: Insights into chemical and biological activities. Quím. Nova 2015, 38, 538–552. [Google Scholar] [CrossRef]
- Goel, A.; Kunnumakkara, A.B.; Aggarwal, B.B. Curcumin as “Curecumin”: From kitchen to clinic. Biochem. Pharm. 2008, 75, 787–809. [Google Scholar] [CrossRef] [Green Version]
- Khor, P.Y.; Aluwi, M.F.F.M.; Rullah, K.; Lam, K.W. Insights on the synthesis of asymmetric curcumin derivatives and their biological activities. Eur. J. Med. Chem. 2019, 183, 111704. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The essential medicinal chemistry of curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Kumar, A.; Bharti, A.C. Anticancer potential of curcumin: Preclinical and clinical studies. Anticancer Res. 2003, 23, 363–398. [Google Scholar] [PubMed]
- Sueth-Santiago, V.; Moraes, J.B.B.; Alves, E.S.S.; Vannier-Santos, M.A.; Freire-de-Lima, C.G.; Castro, R.N.; Mendes-Silva, G.P.; Del Cistia, C.N.; Magalhães, L.G.; Andricopulo, A.D.; et al. The effectiveness of natural diarylheptanoids against Trypanosoma cruzi: Cytotoxicity, ultrastructural alterations and molecular modeling studies. PLoS ONE 2016, 11, e0162926. [Google Scholar] [CrossRef]
- Bland, A.R.; Bower, R.L.; Nimick, M.; Hawkins, B.C.; Rosengren, R.J.; Ashton, J.C. Cytotoxicity of curcumin derivatives in ALK positive non-small cell lung cancer. Eur. J. Pharmacol. 2019, 865, 172749. [Google Scholar] [CrossRef]
- Tan, K.L.; Ali, A.; Du, Y.; Fu, H.; Jin, H.X.; Chin, T.M.; Khan, M.; Go, M.L. Synthesis and evaluation of bisbenzylidenedioxotetrahydrothiopranones as activators of endoplasmic reticulum (ER) stress signaling pathways and apoptotic cell death in acute promyelocytic leukemic cells. J. Med. Chem. 2014, 57, 5904–5918. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Zhang, S.; Zhou, Y.; Zhu, M.; Kang, Y.; Chen, D.; Wang, J.; Zhou, P.; Li, W.; Xu, Q.; et al. Synthesis and evaluation of asymmetric curcuminoid analogs as potential anticancer agents that down regulate NF-kB activation and enhance the sensitivity of gastric cancer cell lines to irinotecan chemotherapy. Eur. J. Med. Chem. 2017, 139, 917–925. [Google Scholar] [CrossRef]
- Shinzato, T.; Sato, R.; Suzuki, K.; Tomioka, S.; Sogawa, H.; Shulga, S.; Blume, Y.; Kurita, N. Proposal of therapeutic curcumin derivatives for Alzheimer’s disease based on ab initio molecular simulations. Chem. Phys. Lett. 2002, 738, 136883. [Google Scholar] [CrossRef]
- Waranyoupalin, R.; Wongnawa, S.; Wongnawa, M.; Pakawatchai, C.; Panichayupakaranant, P.; Sherdshoopongse, P. Studies on complex formation between curcumin and Hg(II) ion by spectrophotometric method: A new approach to overcome peak overlap. Cent. Eur. J. Chem. 2009, 7, 388–394. [Google Scholar] [CrossRef] [Green Version]
- Priyadarsini, K.I. Photophysics, photochemistry and photobiology of curcumin: Studies from organic solutions, bio-mimetics and living cells. J. Photochem. Photobiol. C 2009, 10, 81–95. [Google Scholar] [CrossRef]
- Ran, C.; Xu, X.; Raymond, S.B.; Ferrara, B.J.; Neal, K.; Bacskai, B.J.; Medarova, Z.; Moore, A. Design, synthesis, and testing of difluoroboron-derivatized curcumins as near infrared probes for in vivo detection of amyloid-β deposits. J. Am. Chem. Soc. 2009, 131, 15257–15261. [Google Scholar] [CrossRef] [Green Version]
- Chaicham, A.; Kulchat, S.; Tumcharern, G.; Tuntulani, T.; Tomapatanaget, B. Synthesis, photophysical properties, and cyanide detection in aqueous solution of BF2-curcumin dyes. Tetrahedron 2010, 66, 6217–6223. [Google Scholar] [CrossRef]
- Margar, S.N.; Rhyman, L.; Ramasami, P.; Sekar, N. Fluorescent difluoroboron-curcumin analogs: An investigation of the electronic structures and photophysical properties. Spectrochim. Acta A 2016, 152, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, H.Y.; Kim, Y.S.; Seo, J.H.; Roh, E.J.; Han, H.; Shin, K.J. Small molecules that protect against β-amyloid-induced cytotoxicity by inhibiting aggregation of β-amyloid. Bioorg. Med. Chem. 2012, 20, 4921–4935. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, H.C.; De Almeida, W.B. Theoretical calculations of1H NMR chemical shifts for nitrogenated compounds in chloroform solution. Chem. Phys. 2020, 528, 110479. [Google Scholar] [CrossRef]
- Soares, B.A.; Firme, C.L.; Maciel, M.A.M.; Kaiser, C.R.; Schilling, E.; Bortoluzzi, A.J. Experimental and NMR theoretical methodology applied to geometric analysis of the bioactive clerodane trans-dehydrocrotonin. J. Braz. Chem. Soc. 2014, 25, 629–638. [Google Scholar]
- Souza, L.G.S.; Almeida, M.C.S.; Lemos, T.L.G.; Ribeiro, P.R.V.; Canuto, K.M.; Braz-Filho, R.; Del Cistia, C.N.; Sant’Anna, C.M.R.; Barreto, F.S.; de Moraes, M.O. Brazoides A-D, New Alkaloids from Justicia gendarussa Burm. F. Species. J. Braz. Chem. Soc. 2017, 28, 1281–1287. [Google Scholar] [CrossRef]
- Lyu, H.; Wang, D.; Cai, L.; Wang, D.-J.; Li, X.-M. Synthesis, photophysical and solvatochromic properties of diacetoxyboron complexes with curcumin derivatives. Spectrochim. Acta A 2019, 220, 117126. [Google Scholar] [CrossRef]
- Balasubramanian, K. Theoretical calculations on the transition energies of the UV-visible spectra of curcumin pigment in turmeric. Ind. J. Chem. A 1991, 30, 61–65. [Google Scholar]
- Patra, D.; Barakat, C. Synchronous fluorescence spectroscopic study of solvatochromic curcumin dye. Spectrochim. Acta A 2011, 70, 1034–1041. [Google Scholar] [CrossRef]
- Przybytek, J.T. High-Purity Solvent Guide, 1st ed.; Burdick & Jackson Laboratories: Muskegon, MI, USA, 1980. [Google Scholar]
- Lakowicz, J.R. ; Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, NY, USA, 2006. [Google Scholar]
- Jacques, P. On the relative contributions of nonspecific and specific interactions to the unusual solvatochromism of a typical merocyanine dye. J. Phys. Chem. 1986, 90, 5535–5539. [Google Scholar] [CrossRef]
- Fujisawa, S.; Atsumi, T.; Ishihara, M.; Kadoma, Y. Cytotoxicity, ROS-generation activity and radical-scavenging activity of curcumin and related compounds. Anticancer Res. 2004, 24, 563–570. [Google Scholar]
- Masuda, T.; Toi, Y.; Bando, H.; Maekawa, T.; Takeda, Y.; Yamaguchi, H. Structural identification of new curcumin dimers and their contribution to the antioxidant mechanism of curcumin. J. Agric. Food Chem. 2002, 50, 2524–2530. [Google Scholar] [CrossRef] [PubMed]
- Kosar, B.; Albayrak, C. Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino)methyl]phenol. Spectrochim. Acta A 2011, 78, 160–167. [Google Scholar] [CrossRef]
- Politzer, P.; Lane, P. A computational study of some nitrofluoromethanes. Struct. Chem. 1990, 1, 159–164. [Google Scholar] [CrossRef]
- Lisboa, C.S.; de Lucas, N.C.; Garden, S.J. Synthesis of novel substituted methoxybenzo[2,3-b]carbazole derivatives via C-H functionalization. Experimental and theoretical characterization of their photophysical properties. Dyes Pig. 2016, 134, 618–632. [Google Scholar] [CrossRef]
- Coppo, R.L.; Zanoni, K.P.S.; Iha, N.Y.M. Unraveling the luminescence of new heteroleptic Ir(III) cyclometalated series. Polyhedron 2019, 163, 161–170. [Google Scholar] [CrossRef]
- Katoh, R.; Suzuki, K.; Furube, A.; Kotani, M.; Tokumaru, K. Fluorescence quantum yield of aromatic hydrocarbon crystals. J. Phys. Chem. C 2009, 113, 2961–2965. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent a | λmax (nm) | Log ε b | λexc (nm) | λem (nm) | ΦF | Stokes Shift (nm) | Dimer Formation |
|---|---|---|---|---|---|---|---|
| Methanol | 220; 275; 380 | 4.19 | 222; 300; 392 | 507 | 0.09 | 115 | No |
| Acetonitrile | 225; 255; 390 | 4.48 | 225; 295; 395 | 504 | 0.23 | 109 | No |
| Toluene | 300; 395 | 4.57 | 304; 396 | 443 | 0.38 | 47 | No |
| Solvent | µ (D) | EHOMO (eV) | ELUMO (eV) | |ΔE| (eV) |
|---|---|---|---|---|
| Methanol | 6.26 | −5.70 | −2.41 | 3.29 |
| Acetonitrile | 6.21 | −5.58 | −2.31 | 3.27 |
| Toluene | 5.46 | −5.59 | −2.25 | 3.34 |
| Mulliken Charge in the Ground State | Mulliken Charge in the Excited State | |||||
|---|---|---|---|---|---|---|
| Position a | Methanol | Acetonitrile | Toluene | Methanol | Acetonitrile | Toluene |
| 1 | −0.194 | −0.203 | −0.218 | −0.194 | −0.203 | −0.218 |
| 2 | 0.236 | 0.234 | 0.285 | 0.236 | 0.234 | 0.285 |
| 3 | −0.185 | −0.189 | −0.192 | −0.185 | −0.189 | −0.192 |
| 4 | −0.193 | −0.197 | −0.185 | −0.193 | −0.197 | −0.185 |
| 5 | 0.163 | 0.158 | 0.172 | 0.163 | 0.158 | 0.172 |
| 6 | −0.270 | −0.279 | −0.281 | −0.270 | −0.279 | −0.281 |
| 7 | 0.357 | 0.372 | 0.365 | 0.357 | 0.372 | 0.365 |
| 8 | 0.278 | 0.302 | 0.317 | 0.278 | 0.302 | 0.317 |
| 9 | −0.189 | −0.201 | −0.190 | −0.189 | −0.201 | −0.190 |
| 10 | −0.184 | −0.191 | −0.193 | −0.184 | −0.191 | −0.193 |
| 1′ | −0.126 | −0.116 | −0.116 | −0.126 | −0.116 | −0.116 |
| 2′ | 0.126 | 0.131 | 0.137 | 0.128 | 0.132 | 0.139 |
| 3′ | −0.185 | −0.182 | −0.174 | −0.182 | −0.181 | −0.174 |
| 4′ | −0.198 | −0.205 | −0.190 | −0.198 | −0.205 | −0.190 |
| 5′ | 0.351 | 0.376 | 0.380 | 0.351 | 0.376 | 0.382 |
| 6′ | −0.198 | −0.207 | −0.190 | −0.196 | −0.206 | −0.189 |
| 7′ | −0.211 | −0.220 | −0.209 | −0.210 | −0.218 | −0.207 |
| 1″ | 0.272 | 0.270 | 0.261 | 0.272 | 0.270 | 0.261 |
| C7-OCH3 | −0.242 | −0.242 | −0.232 | −0.242 | −0.242 | −0.232 |
| C5′-OCH3 | −0.242 | −0.240 | −0.229 | −0.242 | −0.240 | −0.229 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaves, O.A.; Sueth-Santiago, V.; Pinto, D.C.d.A.; Netto-Ferreira, J.C.; Decote-Ricardo, D.; Lima, M.E.F.d. 2-Chloro-4,6-bis{(E)-3-methoxy-4-[(4-methoxybenzyl)oxy]styryl}pyrimidine: Synthesis, Spectroscopic and Computational Evaluation. Molbank 2021, 2021, M1276. https://doi.org/10.3390/M1276
Chaves OA, Sueth-Santiago V, Pinto DCdA, Netto-Ferreira JC, Decote-Ricardo D, Lima MEFd. 2-Chloro-4,6-bis{(E)-3-methoxy-4-[(4-methoxybenzyl)oxy]styryl}pyrimidine: Synthesis, Spectroscopic and Computational Evaluation. Molbank. 2021; 2021(3):M1276. https://doi.org/10.3390/M1276
Chicago/Turabian StyleChaves, Otávio Augusto, Vitor Sueth-Santiago, Douglas Chaves de Alcântara Pinto, José Carlos Netto-Ferreira, Debora Decote-Ricardo, and Marco Edilson Freire de Lima. 2021. "2-Chloro-4,6-bis{(E)-3-methoxy-4-[(4-methoxybenzyl)oxy]styryl}pyrimidine: Synthesis, Spectroscopic and Computational Evaluation" Molbank 2021, no. 3: M1276. https://doi.org/10.3390/M1276
APA StyleChaves, O. A., Sueth-Santiago, V., Pinto, D. C. d. A., Netto-Ferreira, J. C., Decote-Ricardo, D., & Lima, M. E. F. d. (2021). 2-Chloro-4,6-bis{(E)-3-methoxy-4-[(4-methoxybenzyl)oxy]styryl}pyrimidine: Synthesis, Spectroscopic and Computational Evaluation. Molbank, 2021(3), M1276. https://doi.org/10.3390/M1276