
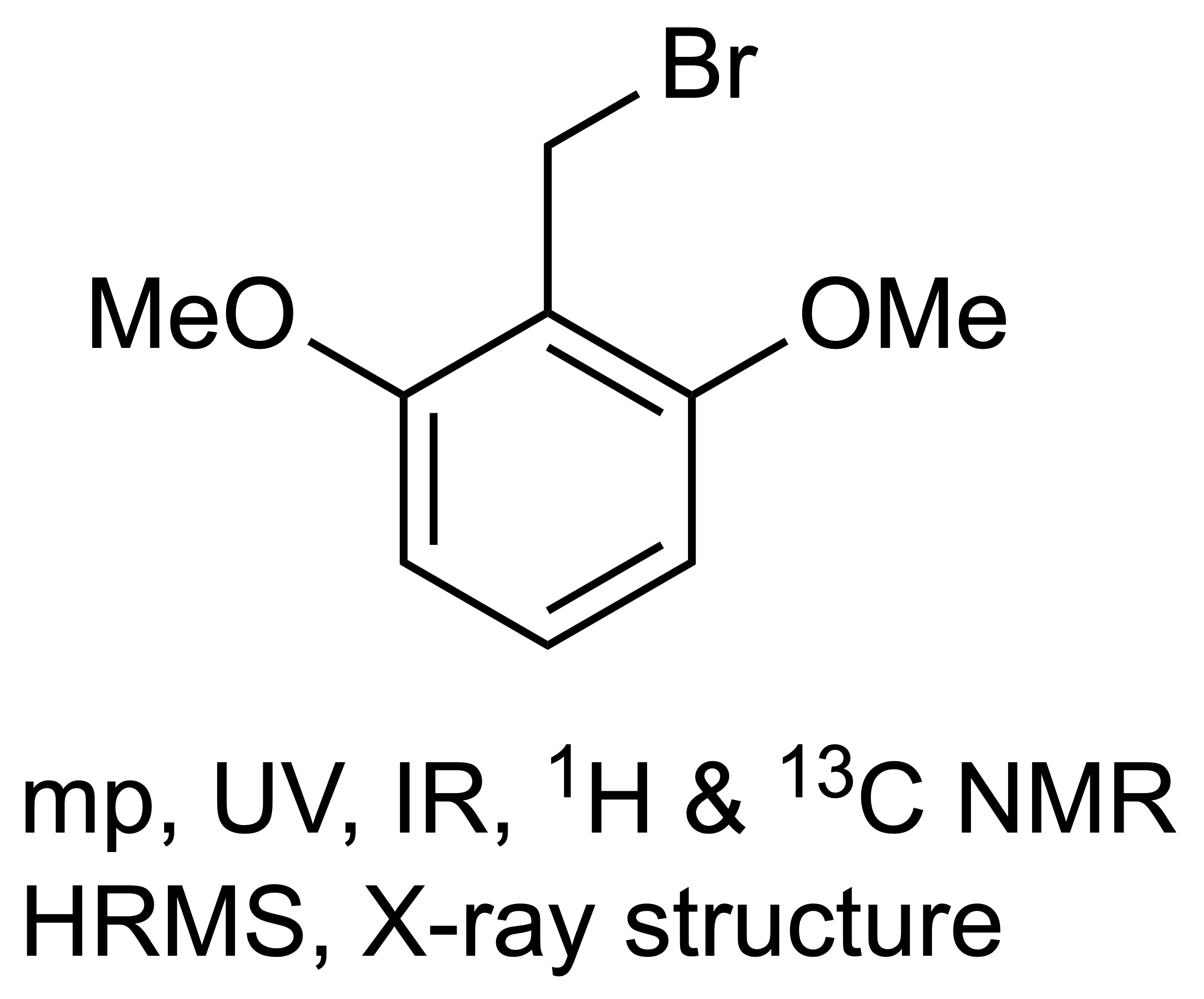
2,6-Dimethoxybenzyl Bromide



Abstract
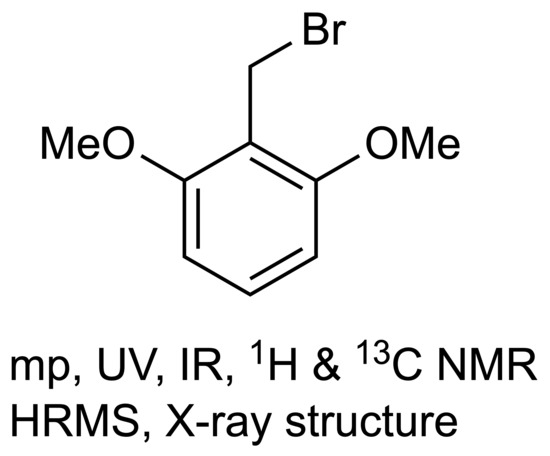
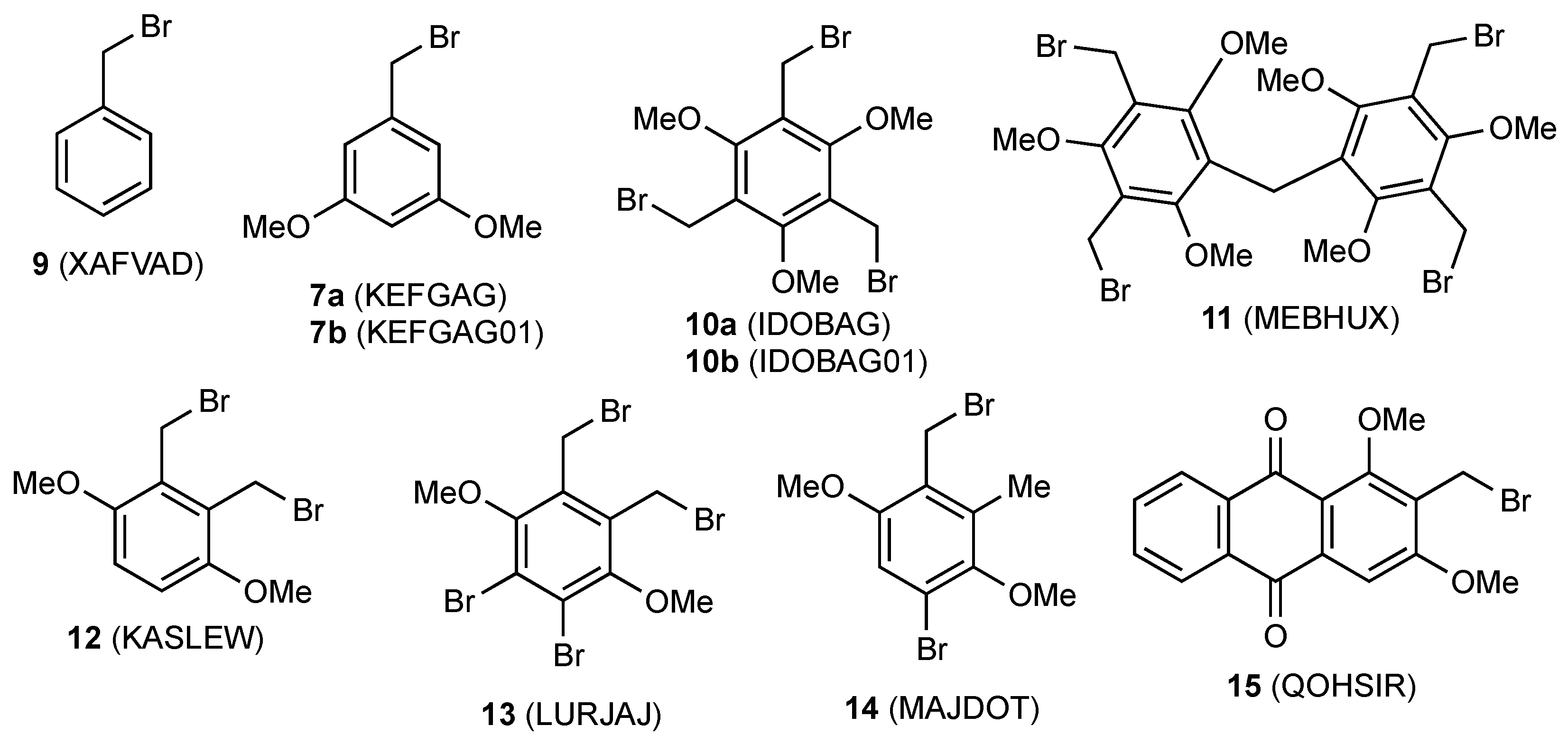
1. Introduction
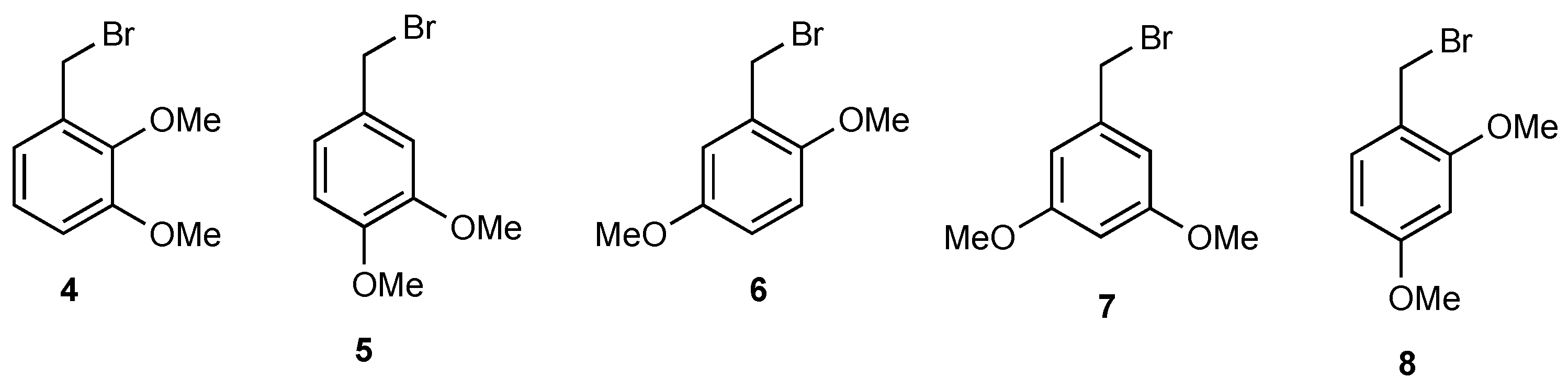
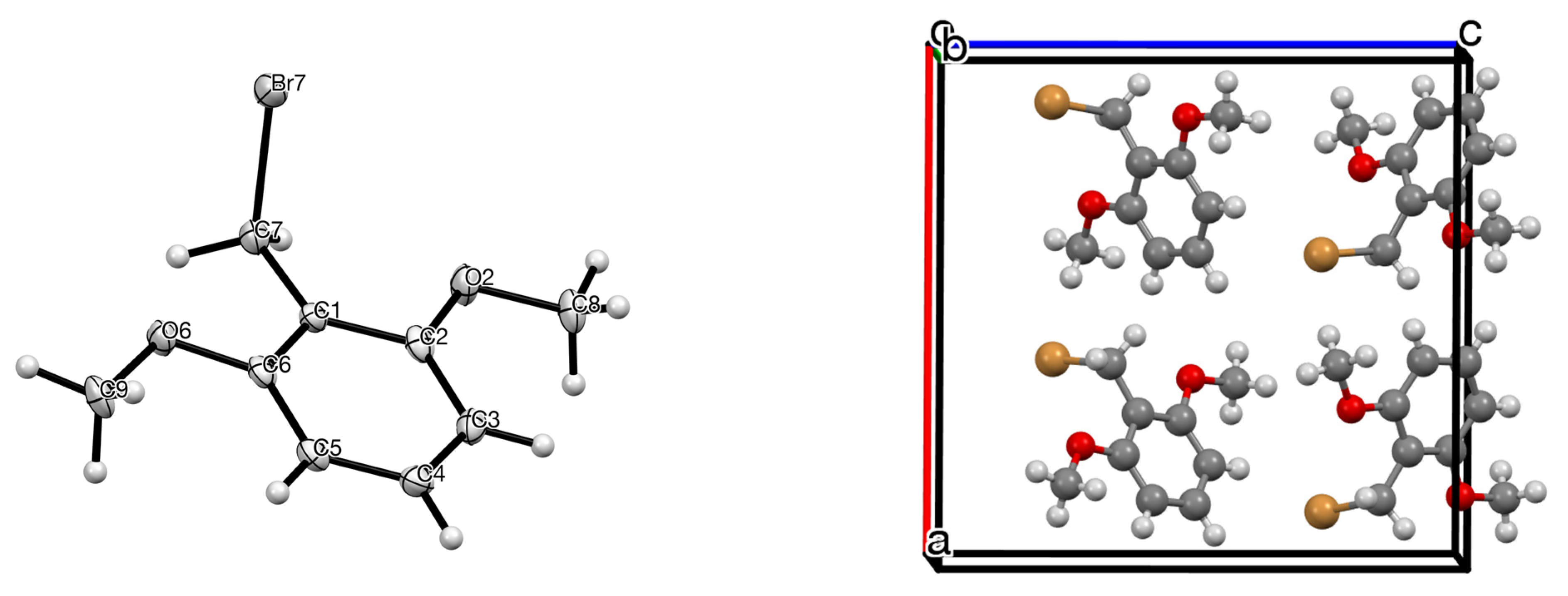
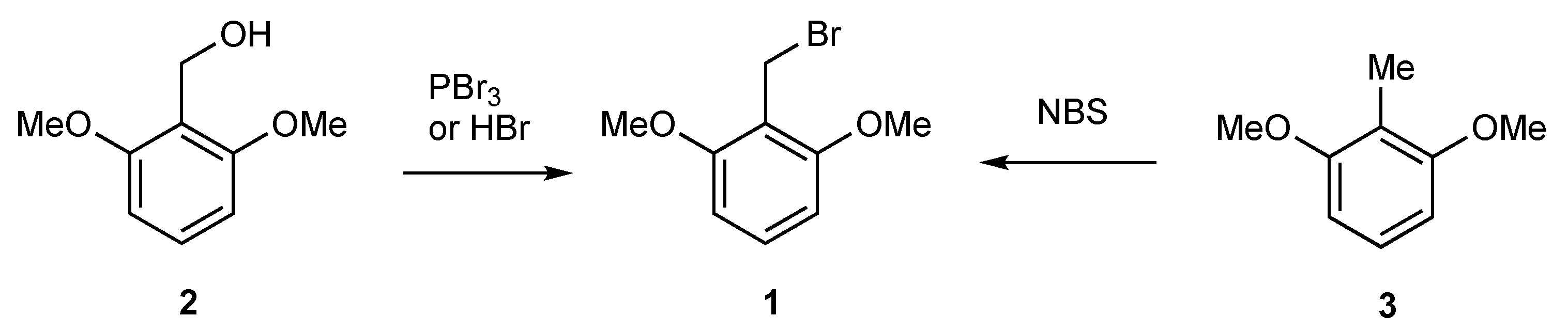
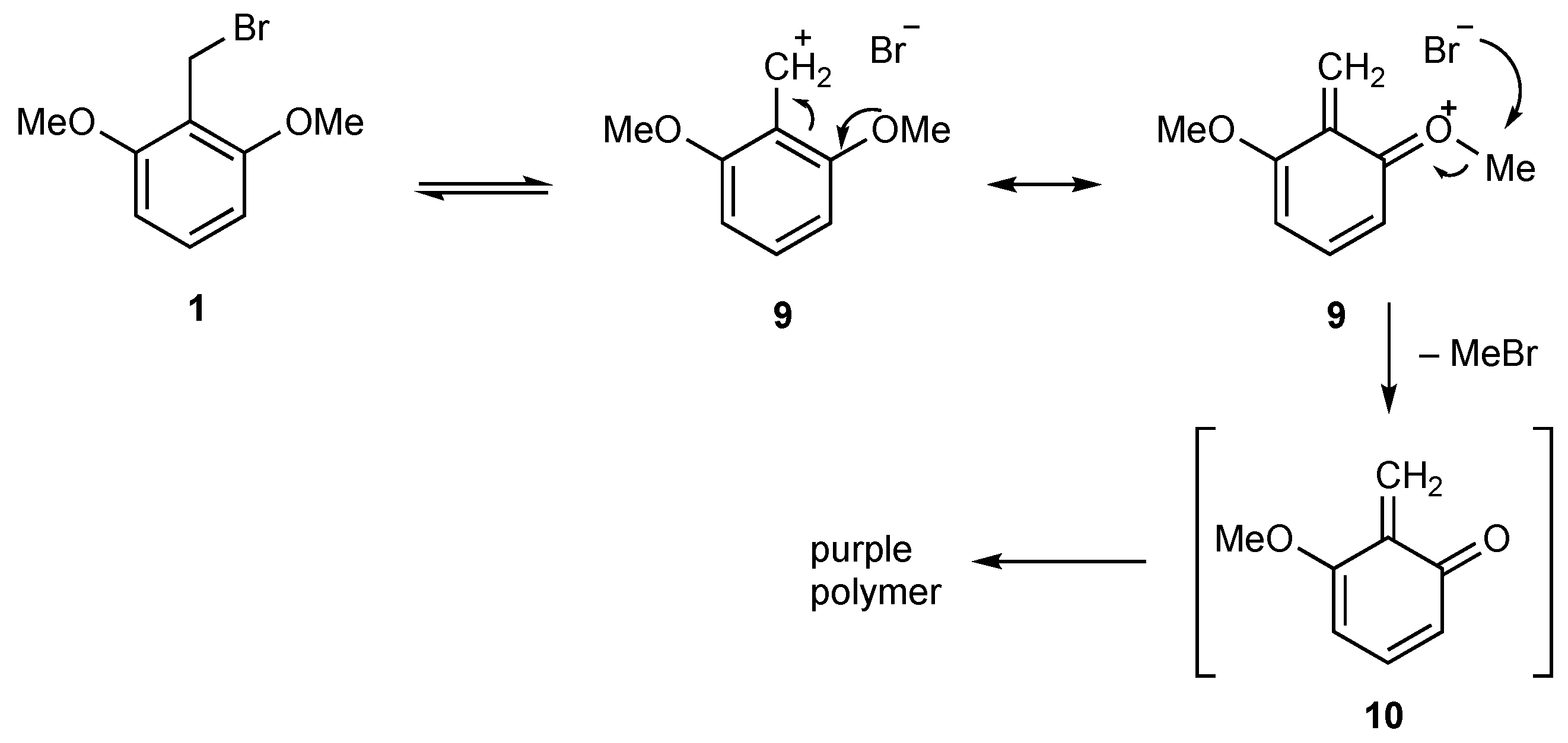
2. Results
3. Experimental Section
2,6-Dimethoxybenzyl Bromide (1)
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nakajima, N.; Abe, R.; Yonemitsu, O. 3-Methoxybenzyl (3-MPM) and 3,5-dimethoxybenzyl (3,5-DMPM) protecting groups for the hydroxy function less readily removable the 4-methoxybenzyl (MPM) and 3,4-dimethoxybenzyl (DMPM) protecting groups by DDQ oxidation. Chem. Pharm. Bull. 1988, 36, 4244–4247. [Google Scholar] [CrossRef]
- Falck, J.R.; Barma, D.K.; Baati, R.; Mioskowski, C. Differential cleavage of arylmethyl ethers: Reactivity of 2,6-dimethoxybenzyl ethers. Angew. Chem. Int. Ed. 2001, 40, 1281–1283. [Google Scholar] [CrossRef]
- Smoot, J.T.; Demchenko, A.V. How the arming participating moieties can broaden the scope of chemoselective oligosaccharide synthesis by allowing the inverse armed-disarmed approach. J. Org. Chem. 2008, 73, 8838–8850. [Google Scholar] [CrossRef] [PubMed]
- Kaloglu, M.; Sahin, N.; Sémeril, D.; Brenner, E.; Matt, D.; Ozdemir, I.; Kaya, C.; Toupet, L. Copper-catalysed allylic substitution using 2,8,14,20-tetrapentylresorcinarenyl-substituted imidazolium salts. Eur. J. Org. Chem. 2015, 7310–7316. [Google Scholar] [CrossRef]
- Bernal, P.; Fernández, R.; Lassaletta, J.M. Organocatalytic asymmetric cyanosilylation of nitroalkenes. Chem. Eur. J. 2010, 16, 7714–7718. [Google Scholar] [CrossRef]
- Feng, L.; Lv, K.; Liu, M.; Wang, S.; Zhao, J.; You, X.; Li, S.; Cao, J.; Guo, H. Synthesis and in vitro antibacterial activity of gemifloxacin derivatives containing a substituted benzyloxime moiety. Eur. J. Med. Chem. 2012, 55, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Yuan, Y.; Liang, C.; Wu, F.; Zhang, S.; Xie, X.; Zhang, Z. Photocatalytic radical ortho-dearomative cyclization: Access to spiro[4.5]deca-1,7,9-trien-6-ones. J. Org. Chem. 2021, 86, 3697–3705. [Google Scholar] [CrossRef]
- Silverman, I.R.; Cohen, D.H.; Lyga, J.W.; Szczepanski, S.W.; Ali, S.F. Insecticidal N-(Substituted Arylmethyl)-4-[(Bis(Substituted Phenyl)Methyl]Piperidines. U.S. Patent 1996 5569664, 29 October 1996. [Google Scholar]
- Nussbaumer, P. Trisubstituted Phenyl Derivatives. U.S. Patent 1999 5990116, 23 November 1999. [Google Scholar]
- Haworth, R.D.; Perkin, W.H., Jr. Synthetical experiments in the isoquinoline group. Part I. J. Chem. Soc. 1925, 127, 1434–1444. [Google Scholar] [CrossRef]
- Haworth, R.D.; Perkin, W.H., Jr.; Rankin, J. Synthetical experiments in the isoquinoline group. Part II. J. Chem. Soc. 1925, 127, 1444–1448. [Google Scholar] [CrossRef]
- Freudenberg, K.; Carrara, G.; Cohn, E. Eine umlagerungsreaktion des catechins. 21. Mitteilung über gerbstoffe und ähnliche verbindungen. Liebigs Ann. Chem. 1926, 446, 87–95. [Google Scholar] [CrossRef]
- Shulgin, A.T.; Gal, E.M. A new synthesis of 2:5-dihydroxyphenyl-DL-alanine adapted to isotopic scale. J. Chem. Soc. 1953, 1316–1318. [Google Scholar] [CrossRef]
- Hartzfeld, D.G.; Roser, S.D. Efficient pyrimidine dimer radical anion splitting in low polarity solvents. J. Am. Chem. Soc. 1993, 115, 850–854. [Google Scholar] [CrossRef]
- Bhati, A. Syntheses of some tetralones related to tetracyclines. Tetrahedron 1962, 18, 1519–1526. [Google Scholar] [CrossRef]
- Pan, Z.; Cheung, E.Y.; Harris, K.D.M.; Constable, E.C.; Housecroft, C.E. Structural properties of methoxy derivatives of benzyl bromide, determined from powder X-ray diffraction data. Powder Diffr. 2005, 20, 345–352. [Google Scholar] [CrossRef][Green Version]
- Flörke, U.; Aamer, S. Experimental crystal structure determination. CSD Commun. 2018, CCDC 1884625. [Google Scholar] [CrossRef]
- Foulon, L.; Garcia, G.; Mettefeu, D.; Serradeil-Legal, C.; Valette, G. 1-Benzyl-1,3-Dihydroindol-2-One Derivatives, Their Preparation and the Pharmaceutical Compositions in which they are Present. U.S. Patent 1997 5618833, 8 April 1997. [Google Scholar]
- Bold, G.; Capraro, H.-G.; Fässler, A.; Lang, M.; Bhagwat, S.S.; Khanna, S.C.; Lazdins, J.K.; Mestan, J. Antiviral Ethers of Aspartate Protease Substrate Isosteres. U.S. Patent 1997 5663200, 2 September 1997. [Google Scholar]
- König, M.; Linhardt, A.; Brüggemenn, O.; Teasdale, I. Phosphine functionalized polyphosphazenes: Soluble and re-usable polymeric reagents for highly efficient halogenations under Appel conditions. Monatsh. Chem. 2016, 147, 1575–1582. [Google Scholar] [CrossRef]
- Bandaranayake, W.M.; Crombie, L.; Whiting, D.A. Pyridine-catalysed chromenylation of mono-chelated meta-dihydric-phenols with mono-, sesqui- and di-terpene aldehydes: Synthesis of rubranine and flemingins A-, B- and C-methyl ethers. J. Chem. Soc. C 1971, 804–810. [Google Scholar] [CrossRef]
- Nayak, S.K.; Sathishkumar, R.; Guru Row, T.N. Directing role of functional groups in selective generation of C–H...π interactions: In situ cryo-crystallographic studies on benzyl derivatives. CrystEngComm 2010, 12, 3112–3118. [Google Scholar] [CrossRef]
- Koch, N.; Seichter, W.; Mazik, M. 1,3,5-Tris(bromomethyl)-2,4,6-trimethoxybenzene. Acta Crystallogr. Sect. E 2013, 69, o679. [Google Scholar] [CrossRef]
- Nielsen, B.E.; Gotfredsen, H.; Rasmusen, B.; Tortzen, C.G.; Pittelkow, M. Simple procedures for the preparation of 1,3,5-trisubstitited 2,4,6-trimethoxybenzenes. Synlett 2013, 24, 2437–2442. [Google Scholar] [CrossRef][Green Version]
- Morgans, G.L.; van Otterlo, W.A.L.; Michael, J.P.; Fernandes, M.A. 1-[3,5-Bis(bromomethyl)-2,4,6-trimethoxybenzyl]-3,5-bis(bromomethyl)-2,4,6-trimethoxybenzene. Acta Crystallogr. Sect. E 2006, 62, o168–o170. [Google Scholar] [CrossRef]
- Hammerhøj, P.; Christensen, J.B. 2,3-Bis(bromomethyl)-1,4-dimethoxybenzene. Acta Crystallogr. Sect. E 2005, 61, o2839–o2840. [Google Scholar] [CrossRef]
- Mendez-Rojas, M.A.; Ejsmont, K.; Watson, W.H. Synthesis and characterization of a 5,6-dibromobenzoquinone-phenyl maleimide adduct. J. Chem. Crystallogr. 2002, 32, 177–184. [Google Scholar] [CrossRef]
- Aitken, R.A.; Jethwa, S.J.; Richardson, N.V.; Slawin, A.M.Z. Regioselective bromination of 1,4-dimethoxy-2,3-dimethylbenzene and conversion into sulfur-functionalised benzoquinones. Tetrahedron Lett. 2016, 57, 1563–1566. [Google Scholar] [CrossRef]
- Akhtar, M.N.; Zareen, S.; Yeap, S.K.; Ho, W.Y.; Lo, K.M.; Hasan, A.; Alitheen, N.B. Total synthesis, cytotoxic effects of damnacanthal, nordamnacanthal and related anthraquinone analogues. Molecules 2013, 18, 10042–10055. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELXL. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | Length C–CH2 (Å) | Length CH2–Br (Å) | Angle C–CH2–Br ° | Torsion Angle Ring/CH2–Br ° | Ref |
|---|---|---|---|---|---|
| 1 | 1.488 (6) | 1.992 (4) | 112.4 (3) | 88.67 | this work |
| 9 | 1.465 (14) | 1.971 (11) | 110.3 (6) | 77.07 | [22] |
| 7a a | 1.466 (2) | 1.908 (1) | 116.68 (8) | 75.65 | [16] |
| 7b | 1.498 (4) | 1.987 (3) | 110.8 (2) | 75.82 | [17] |
| 10a b | 1.475 (4)–1.491 (5) | 1.964 (4)–1.980 (2) | 111.2 (2)–113.4 (2) | 78.57–89.93 | [23] |
| 10b c | 1.485 (4)–1.498 (6) | 1.972 (4)–1.988 (3) | 110.7 (2)–114.2 (2) | 76.64–89.86 | [24] |
| 11 | 1.495 (3) | 1.979 (3) | 111.6 (2) | 89.38 | [25] |
| 1.491 (3) | 1.984 (2) | 113.1 (2) | 83.19 | ||
| 12 | 1.491 (3) | 1.987 (2) | 112.0 (1) | 83.43 | [26] |
| 1.493 (2) | 1.978 (2) | 111.2 (1) | 79.42 | ||
| 13 | 1.49 (1) | 1.947 (8) | 111.4 (5) | 81.47 | [27] |
| 1.50 (1) | 1.965 (8) | 110.4 (5) | 84.45 | ||
| 14 | 1.49 (1) | 1.991 (7) | 112.3 (5) | 82.45 | [28] |
| 15 | 1.497 (4) | 1.975 (3) | 110.1 (2) | 78.99 | [29] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aitken, R.A.; Saab, E.A.; Slawin, A.M.Z. 2,6-Dimethoxybenzyl Bromide. Molbank 2021, 2021, M1277. https://doi.org/10.3390/M1277
Aitken RA, Saab EA, Slawin AMZ. 2,6-Dimethoxybenzyl Bromide. Molbank. 2021; 2021(3):M1277. https://doi.org/10.3390/M1277
Chicago/Turabian StyleAitken, R. Alan, Elizabeth A. Saab, and Alexandra M. Z. Slawin. 2021. "2,6-Dimethoxybenzyl Bromide" Molbank 2021, no. 3: M1277. https://doi.org/10.3390/M1277
APA StyleAitken, R. A., Saab, E. A., & Slawin, A. M. Z. (2021). 2,6-Dimethoxybenzyl Bromide. Molbank, 2021(3), M1277. https://doi.org/10.3390/M1277