
1-Tosyl-6-vinyl-4,5,6,7-tetrahydro-1H-benzo [d] imidazole-2-amine


{kind=link}
{kind=link}
{kind=link}
Abstract
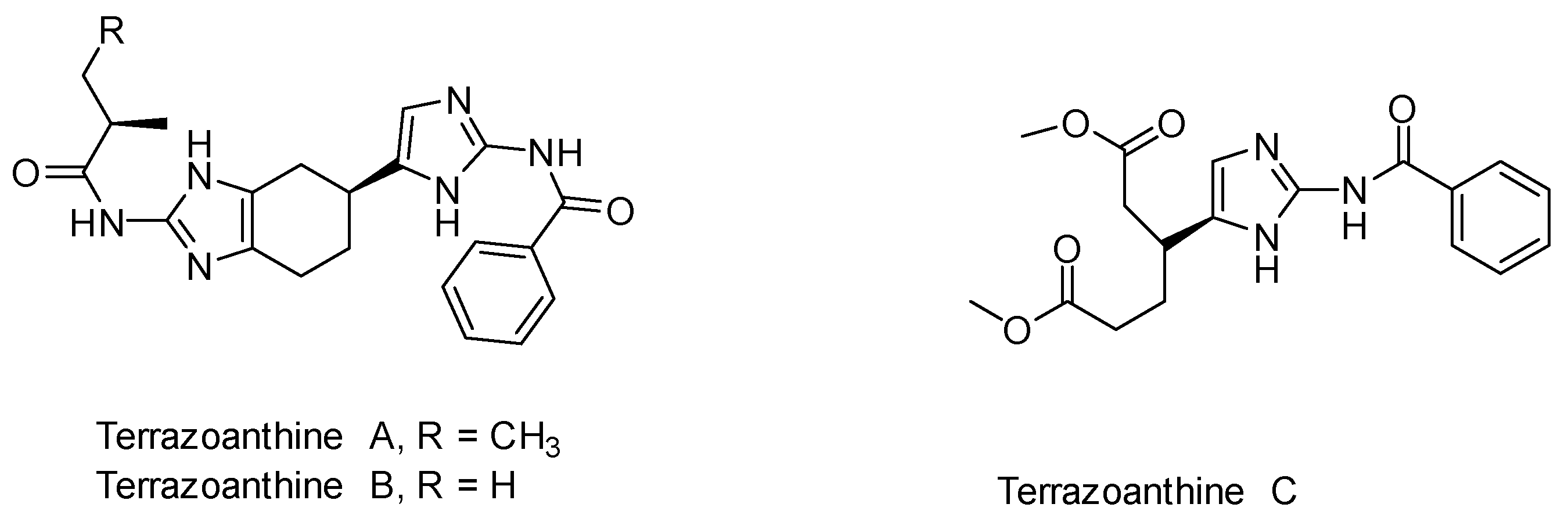
:1. Introduction
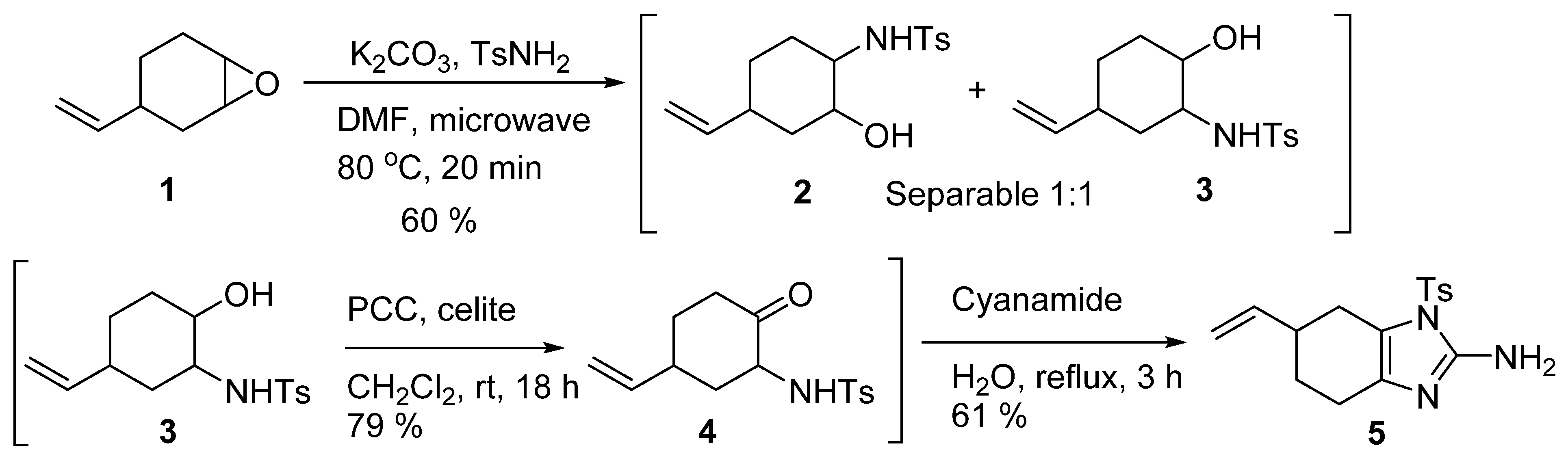
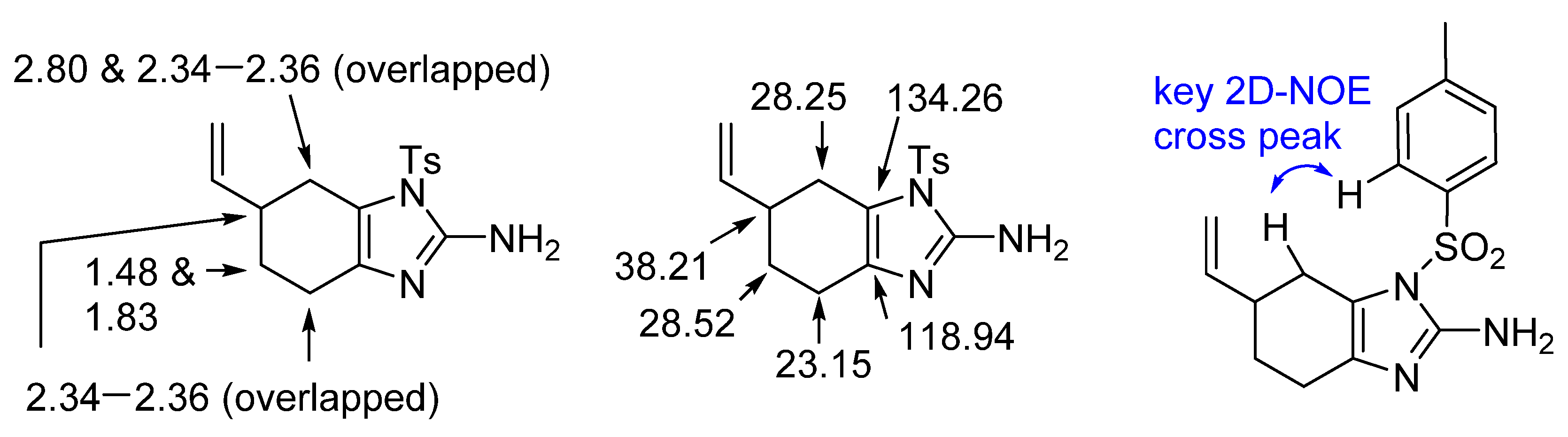
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. Tosyl-6-vinyl-4,5,6,7-tetrahydro-1 H-benzo[d]imidazole-2-amine (5)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guillen, P.O.; Jaramillo, K.B.; Genta-Jouve, G.; Sinniger, F.; Rodriguez, J.; Thomas, O.P. Terrazoanthines, 2-Aminoimidazole Alkaloids from the Tropical Eastern Pacific Zoantharian Terrazoanthus Onoi. Org. Lett. 2017, 19, 1558–1561. [Google Scholar] [CrossRef] [Green Version]
- Arita, K.; Hashimoto, H.; Shimizu, T.; Nakashima, K.; Yamada, M.; Sato, M. Structural Basis for Ca(2+)-Induced Activation of Human PAD4. Nat. Struct. Mol. Biol. 2004, 11, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Pitts, W.J.; Wityak, J.; Smallheer, J.M.; Tobin, A.E.; Jetter, J.W.; Buynitsky, J.S.; Harlow, P.P.; Solomon, K.A.; Corjay, M.H.; Mousa, S.A.; et al. Isoxazolines as Potent Antagonists of the Integrin αvβ3. J. Med. Chem. 2000, 43, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Huigens, R.W.; Reyes, S.; Reed, C.S.; Bunders, C.; Rogers, S.A.; Steinhauer, A.T.; Melander, C. The Chemical Synthesis and Antibiotic Activity of a Diverse Library of 2-Aminobenzimidazole Small Molecules against MRSA and Multidrug-Resistant A. Baumannii. Bioorganic Med. Chem. 2010, 18, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.A.; Bero, J.D.; Melander, C. Chemical Synthesis and Biological Screening of 2-Aminoimidazole-Based Bacterial and Fungal Antibiofilm Agents. ChemBioChem 2010, 11, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Baran, P.S.; Zografos, A.L.; O’Malley, D.P. Short Total Synthesis of (±)-Sceptrin. J. Am. Chem. Soc. 2004, 126, 3726–3727. [Google Scholar] [CrossRef] [PubMed]
- Baran, P.S.; Li, K.; O’Malley, D.P.; Mitsos, C. Short, Enantioselective Total Synthesis of Sceptrin and Ageliferin by Programmed Oxaquadricyclane Fragmentation. Angew. Chem. Int. Ed. 2005, 45, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Baran, P.S.; O’Malley, D.P.; Zografos, A.L. Sceptrin as a Potential Biosynthetic Precursor to Complex Pyrrole-Imidazole Alkaloids: The Total Synthesis of Ageliferin. Angew. Chem. Int. Ed. 2004, 43, 2674–2677. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.K.; Sarpong, R.; Stoltz, B.M. The First Total Synthesis of Dragmacidin D Protocols from a Number of Deep Water Sponges Including. J. Am. Chem. Soc. 2002, 124, 13179–13184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, N.K.; Caspi, D.D.; Stoltz, B.M. The Total Synthesis of (+)-Dragmacidin F. J. Am. Chem. Soc. 2004, 126, 9552–9553. [Google Scholar] [CrossRef] [PubMed]
- Little, T.L.; Webber, S.E. A Simple and Practical Synthesis of 2-Aminoimidazoles. J. Org. Chem. 1994, 59, 7299–7305. [Google Scholar] [CrossRef]
- Sullivan, J.; Giles, R.; Looper, R. 2-Aminoimidazoles from Leucetta Sponges: Synthesis and Biology of an Important Pharmacophore. Curr. Bioact. Compd. 2009, 5, 39–78. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.M.; Murray, W.V. Microwave Assisted Ring-Opening of Epoxides with N-Biaryl Sulfonamides in the Synthesis of Matrix Metalloproteinase-9 Inhibitors. Tetrahedron Lett. 2008, 49, 835–839. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meany, F.B.; O’Rourke, S.; Murphy, P.V. 1-Tosyl-6-vinyl-4,5,6,7-tetrahydro-1H-benzo [d] imidazole-2-amine. Molbank 2021, 2021, M1262. https://doi.org/10.3390/M1262
Meany FB, O’Rourke S, Murphy PV. 1-Tosyl-6-vinyl-4,5,6,7-tetrahydro-1H-benzo [d] imidazole-2-amine. Molbank. 2021; 2021(3):M1262. https://doi.org/10.3390/M1262
Chicago/Turabian StyleMeany, Fiach B., Sarah O’Rourke, and Paul V. Murphy. 2021. "1-Tosyl-6-vinyl-4,5,6,7-tetrahydro-1H-benzo [d] imidazole-2-amine" Molbank 2021, no. 3: M1262. https://doi.org/10.3390/M1262
APA StyleMeany, F. B., O’Rourke, S., & Murphy, P. V. (2021). 1-Tosyl-6-vinyl-4,5,6,7-tetrahydro-1H-benzo [d] imidazole-2-amine. Molbank, 2021(3), M1262. https://doi.org/10.3390/M1262