
Bioactive Antidiabetic Flavonoids from the Stem Bark of Cordia dichotoma Forst.: Identification, Docking and ADMET Studies


 , , , and
, , , and
Abstract
1. Introduction
2. Results and Discussion
2.1. Identification of Isolated Phytocompounds
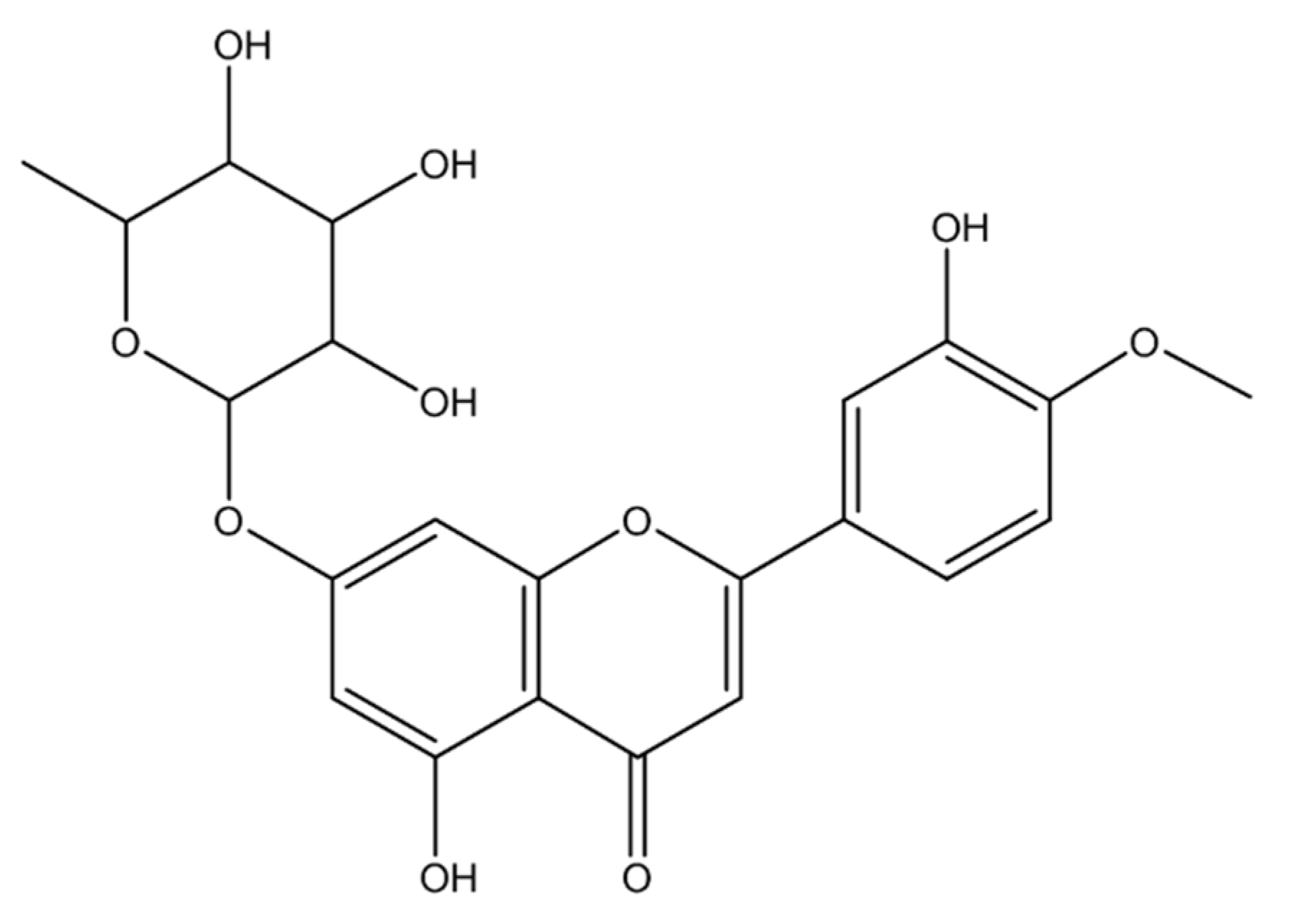
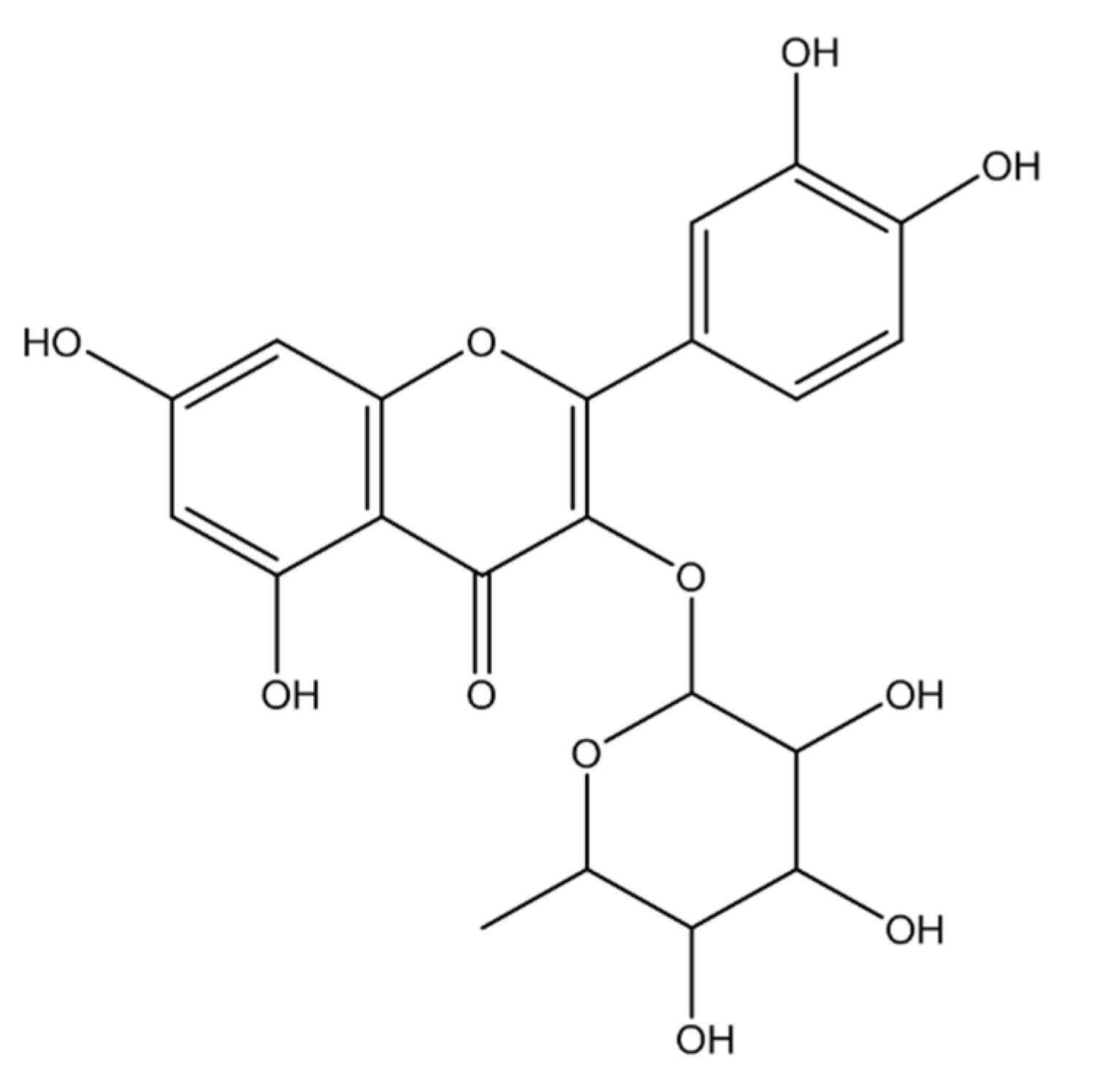
2.1.1. Compound 1 (MECD-1)
2.1.2. Compound 2 (MECD-2)
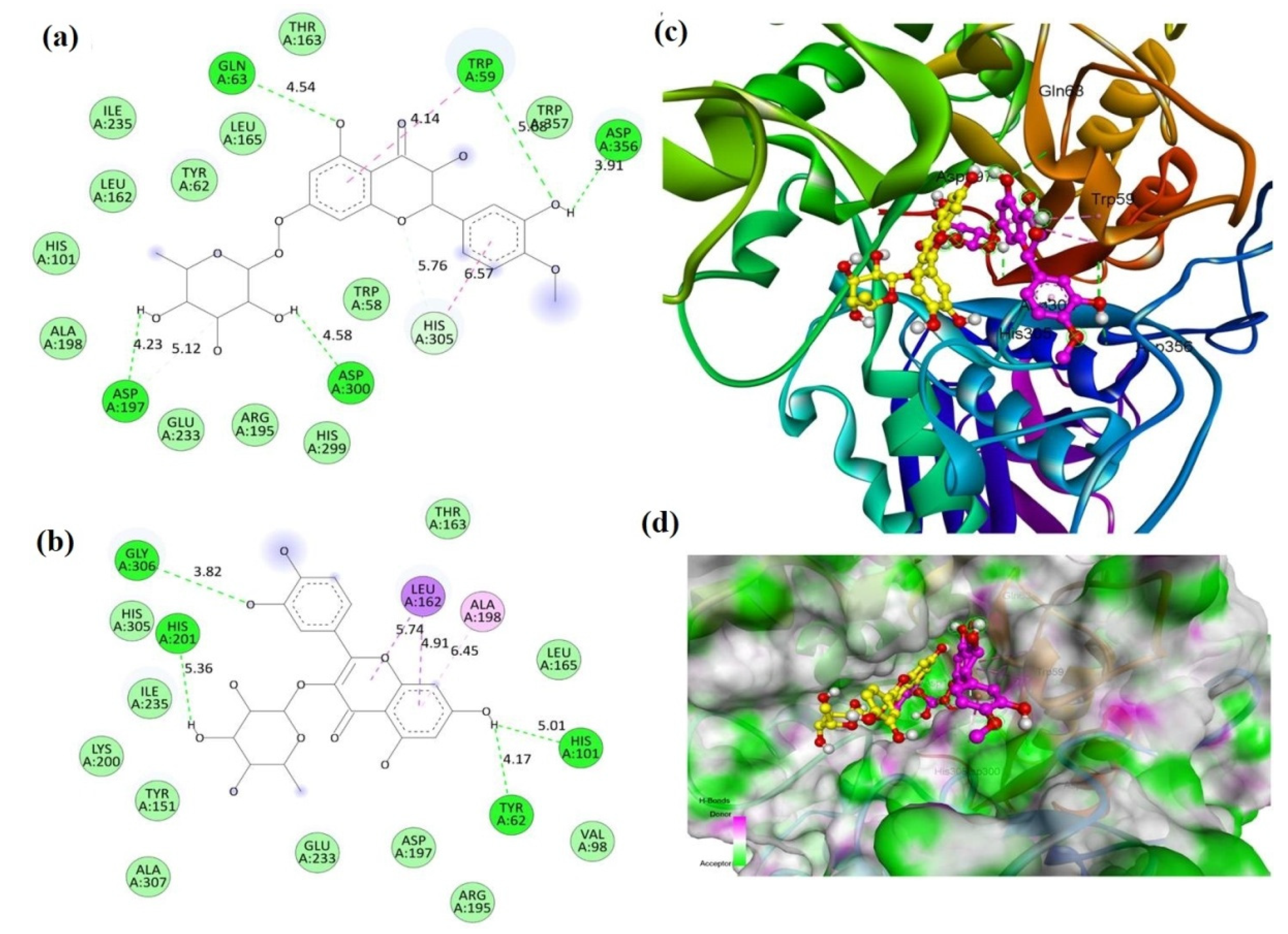
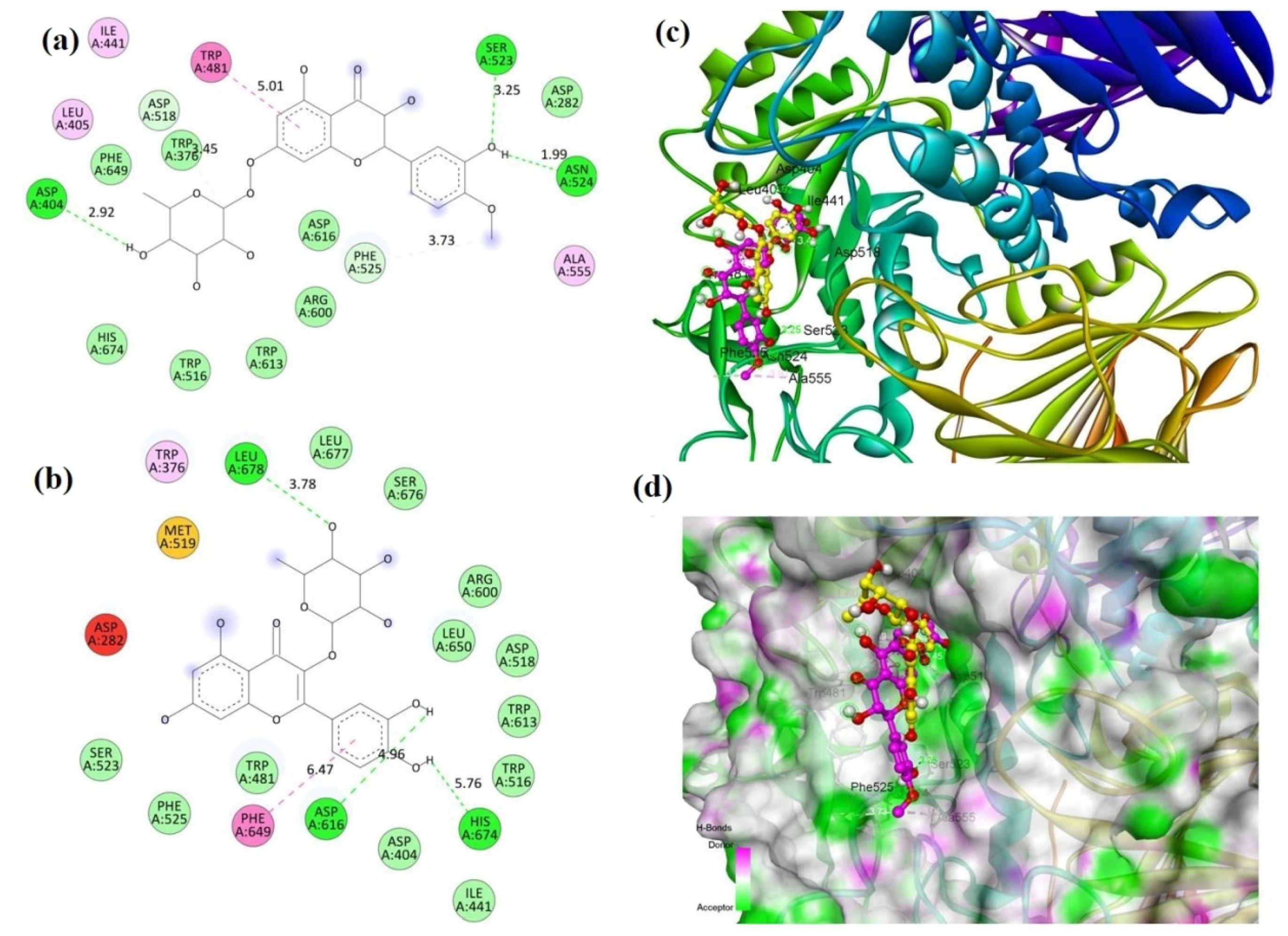
2.2. Molecular Docking
2.3. ADMET
3. Materials and Methods
3.1. Collection of Plant
3.2. Preparation of Methanolic Bark Extract
3.3. Phytochemical Analysis
3.4. Isolation of Phytocompounds
3.5. Identification of Isolated Compounds
3.6. Molecular Docking
3.7. ADMET Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jamkhande, P.G.; Barde, S.R.; Patwekar, S.L.; Tidke, P.S. Plant profile, phytochemistry and pharmacology of Cordia dichotoma (Indian cherry): A review. Asian Pac. J. Trop. Biomed. 2013, 3, 1009–1012. [Google Scholar] [CrossRef]
- Hussain, N.; Kakoti, B.B.; Rudrapal, M.; Junejo, J.A.; Laskar, M.A.; Lal, M.; Sarwa, K.K. Anticancer and Antioxidant Activities of Cordia dichotoma Forst. Int. J. Green. Pharm. 2020, 14, 265–273. [Google Scholar]
- Hussain, N.; Kakoti, B.B.; Rudrapal, M.; Sarwa, K.K. Anti-inflammatory and Antioxidant Activities of Cordia dichotoma Forst. Biomed. Pharmacol. J. 2020, 13, 2093–2099. [Google Scholar] [CrossRef]
- Hussain, N.; Kakoti, B.B.; Rudrapal, M.; Rahman, Z.; Rahman, M.; Chutia, D.; Sarwa, K.K. Antidiabetic Activity of the bark of Indian Cherry, Cordia dichotoma. Biosci. Biotech. Res. Comm. 2020, 13, 2211–2216. [Google Scholar] [CrossRef]
- Ragasa, C.Y.; EbajoJr, V.D.; Mariquit, M.; Mandia, E.H.; Tan, M.C.S.; Brkljača, R.; Urban, S. Chemical constituents of Cordia dichotoma G. Forst. J. Appl. Pharm. Sci. 2015, 5, 16–21. [Google Scholar]
- Junejo, J.A.; Rudrapal, M.; Mohammed, A.; Zaman, K. New flavonoid with antidiabetic potential from Tetrastigma angustifolia (Roxb.) Deb leaves. Braz. J. Pharm. Sci. 2020, 56, e18806. [Google Scholar] [CrossRef]
- Junejo, J.A.; Mondal, P.; Rudrapal, M.; Zaman, K. Antidiabetic assessment of the hydro-alcoholicleaf extracts of the plant Tetrastigma angustifolia (Roxb.), a traditionally used North-Eastern Indian vegetable. Biomed. Pharmacol. J. 2014, 7, 635–644. [Google Scholar] [CrossRef]
- Junejo, J.A.; Gogoi, G.; Islam, J.; Rudrapal, M.; Mondal, P.; Hazarika, H.; Zaman, K. Exploration ofantioxidant, antidiabetic and hepatoprotective activity of Diplazium esculentum, a wild edible plant from North Eastern region of India. Future J. Pharm. Sci. 2018, 4, 93–101. [Google Scholar] [CrossRef]
- Kumar, S.; Kaushik, A.; Narasimhan, B.; Shah, S.A.; Lim, S.M.; Ramasamy, K.; Mani, V. Molecular docking, synthesis and biological significance of pyrimidine analogues as prospective antimicrobial and antiproliferative agents. BMC Chem. 2019, 13, 85. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Alseekh, S.; Fernie, A.R. Conservation and diversification of flavonoid metabolism in the plant kingdom. Curr. Opin. Plant. Biol. 2020, 55, 100–108. [Google Scholar] [CrossRef]
- Arora, S.; Itankar, P. Extraction, isolation and identification of flavonoid from Chenopodium album aerial parts. J. Trad. Comp. Med. 2018, 8, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Kishore, N.; Twilley, D.; Blom van Staden, A.; Verma, P.; Singh, B.; Cardinali, G.; Lall, N. Isolation of flavonoids and flavonoid glycosides from Myrsine africana and their inhibitory activities against mushroom tyrosinase. J. Nat. Prod. 2018, 81, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Tundis, R.; Loizzo, M.R.; Menichini, F. Natural products as α-amylase and α-glucosidase inhibitors and their hypoglycaemic potential in the treatment of diabetes: An update. Mini Rev. Med. Chem. 2010, 10, 315–331. [Google Scholar] [CrossRef]
- Junejo, J.A.; Zaman, K.; Rudrapal, M.; Hussain, N. Antidiabetic and Antioxidant Activity of Hydro-alcoholic Extract of Oxalis debilis Kunth. Leaves in Experimental Rats. Biosci. Biotech. Res. Comm. 2020, 13, 860–867. [Google Scholar] [CrossRef]
- Junejo, J.A.; Rudrapal, M.; Zaman, K. Antidiabetic activity of Carallia brachiata Lour. Leaves hydro-alcoholic extract (HAE) with antioxidant potential in diabetic rats. Indian J. Nat. Prod. Resour. 2020, 11, 18–29. [Google Scholar]
- Junejo, J.A.; Rudrapal, M.; Nainwal, L.M.; Zaman, K. Antidiabetic activity of hydro-alcoholic stem bark extract of Callicarpa arborea Roxb. with antioxidant potential in diabetic rats. Biomed. Pharmacother. 2017, 95, 84–94. [Google Scholar] [CrossRef]
- Rashed, K.N.; Butnariu, M. Isolation and antimicrobial and antioxidant evaluation of bioactive compounds from Eriobotrya japonica stem. Adv. Pharm. Bull. 2014, 4, 75–81. [Google Scholar]
- Conforti, F.; Statti, G.A.; Tundis, R.; Menichini, F.; Houghton, P. Antioxidant activity of methanolic extract of Hypericum triquetrifolium Turra aerial part. Fitoterapia 2002, 73, 479–483. [Google Scholar] [CrossRef]
- Lynch, T.; Price, A.L. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician. 2007, 76, 391–396. [Google Scholar]
- Feng, B.; Mills, J.B.; Davidson, R.E.; Mireles, R.J.; Janiszewski, J.S.; Troutman, M.D.; de Morais, S.M. In vitro P-glycoprotein assays to predict the in vivo interactions of P-glycoprotein with drugs in the central nervous system. Drug Metab. Dispos. 2008, 36, 268–275. [Google Scholar] [CrossRef]
- Isyaku, Y.; Uzairu, A.; Uba, S. Computational studies of a series of 2-substituted phenyl-2-oxo-,2-hydroxyl-and 2-acylloxyethylsulfonamides as potent anti-fungal agents. Heliyon 2020, 6, e03724. [Google Scholar] [CrossRef]
- Onah, O.E.; Babangida, K.J. Phytochemical Investigation and Antimicrobial Activity of Hexane, Ethyl Acetate and Methanol Fractions from Stem Bark of Icacina trichantha Oliv (Icacinaceae). J. Chem. Environ. Sci. Appl. 2020, 7, 7–12. [Google Scholar] [CrossRef]
- Jigna, P.; Sumitra, C. Phytochemical screening of some plants from western region of India. Plan. Arch. 2008, 8, 657–662. [Google Scholar]
- Roig-Zamboni, V.; Cobucci-Ponzano, B.; Iacono, R.; Ferrara, M.C.; Germany, S.; Bourne, Y.; Sulzenbacher, G. Structure of human lysosomal acid α-glucosidase–a guide for the treatment of Pompe disease. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nahoum, V.; Roux, G.; Anton, V.; Rougé, P.; Puigserver, A.; Bischoff, H.; Payan, F. Crystal structures of human pancreatic α-amylase in complex with carbohydrate and proteinaceous inhibitors. Biochem. J. 2000, 346, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDockVina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multi threading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. Swiss ADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Kato-Schwartz, C.G.; deSá-Nakanishi, A.B.; Guidi, A.C.; Gonçalves, D.A.; Bueno, F.G.; Zani, B.P.M.; Peralta, R.M. Carbohydrate digestive enzymes are inhibited by Poincianellapluviosa stem bark extract: Relevance on type 2 diabetes treatment. Clin. Phytosci. 2020, 6, 1–11. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Binding Energy (kcal/mole) | |
|---|---|---|
| 5NN8 | 1B2Y | |
| Compound 1 (MECD-1) | −7.8 | −8.6 |
| Compound 2 (MECD-2) | −8.0 | −7.8 |
| Acarbose (Standard drug) | −7.6 | −9.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, N.; Kakoti, B.B.; Rudrapal, M.; Sarwa, K.K.; Celik, I.; Attah, E.I.; Khairnar, S.J.; Bhattacharya, S.; Sahoo, R.K.; Walode, S.G. Bioactive Antidiabetic Flavonoids from the Stem Bark of Cordia dichotoma Forst.: Identification, Docking and ADMET Studies. Molbank 2021, 2021, M1234. https://doi.org/10.3390/M1234
Hussain N, Kakoti BB, Rudrapal M, Sarwa KK, Celik I, Attah EI, Khairnar SJ, Bhattacharya S, Sahoo RK, Walode SG. Bioactive Antidiabetic Flavonoids from the Stem Bark of Cordia dichotoma Forst.: Identification, Docking and ADMET Studies. Molbank. 2021; 2021(2):M1234. https://doi.org/10.3390/M1234
Chicago/Turabian StyleHussain, Nazim, Bibhuti Bhushan Kakoti, Mithun Rudrapal, Khomendra Kumar Sarwa, Ismail Celik, Emmanuel Ifeanyi Attah, Shubham Jagadish Khairnar, Soumya Bhattacharya, Ranjan Kumar Sahoo, and Sanjay G. Walode. 2021. "Bioactive Antidiabetic Flavonoids from the Stem Bark of Cordia dichotoma Forst.: Identification, Docking and ADMET Studies" Molbank 2021, no. 2: M1234. https://doi.org/10.3390/M1234
APA StyleHussain, N., Kakoti, B. B., Rudrapal, M., Sarwa, K. K., Celik, I., Attah, E. I., Khairnar, S. J., Bhattacharya, S., Sahoo, R. K., & Walode, S. G. (2021). Bioactive Antidiabetic Flavonoids from the Stem Bark of Cordia dichotoma Forst.: Identification, Docking and ADMET Studies. Molbank, 2021(2), M1234. https://doi.org/10.3390/M1234