
1-Methyl-8-phenyl-1,3-diazaspiro[4.5]decane-2,4-dione




Abstract
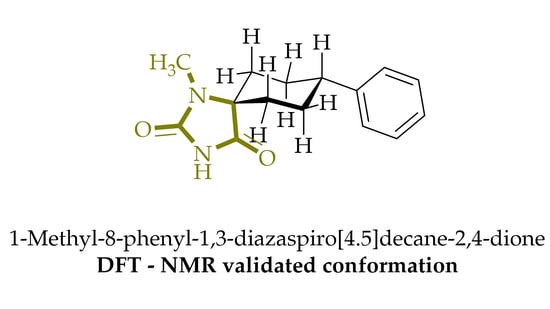
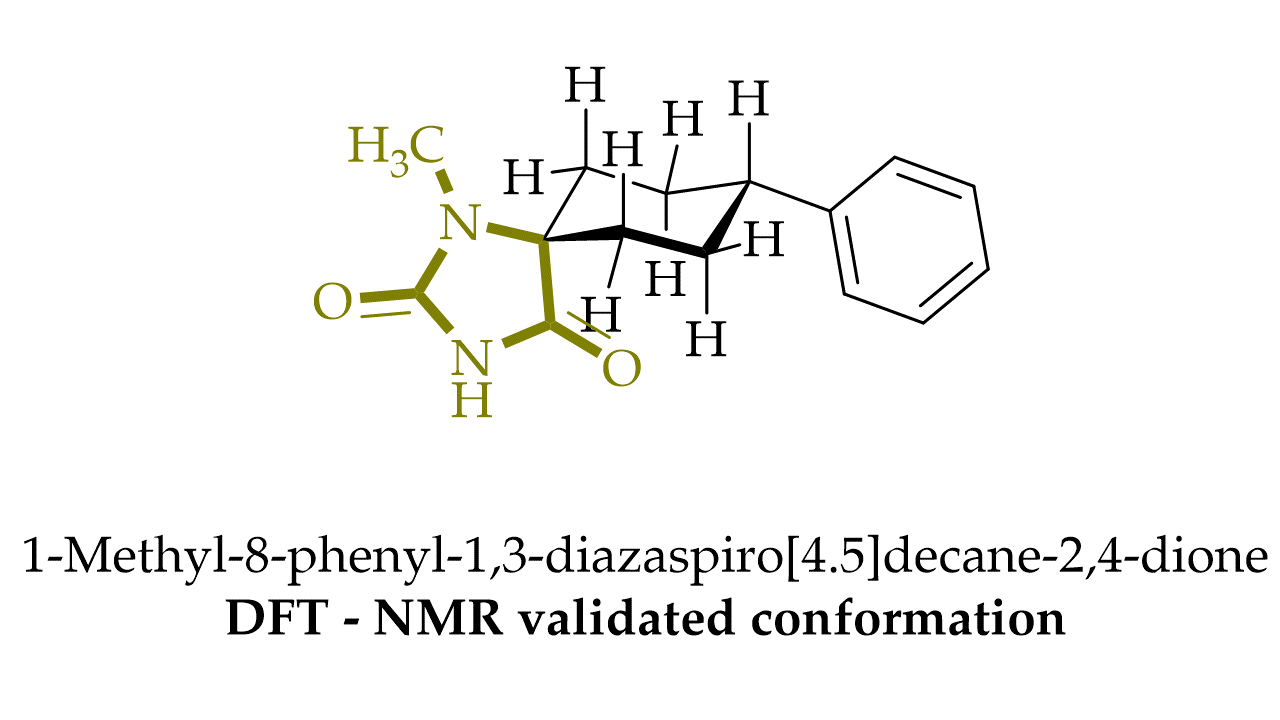
1. Introduction
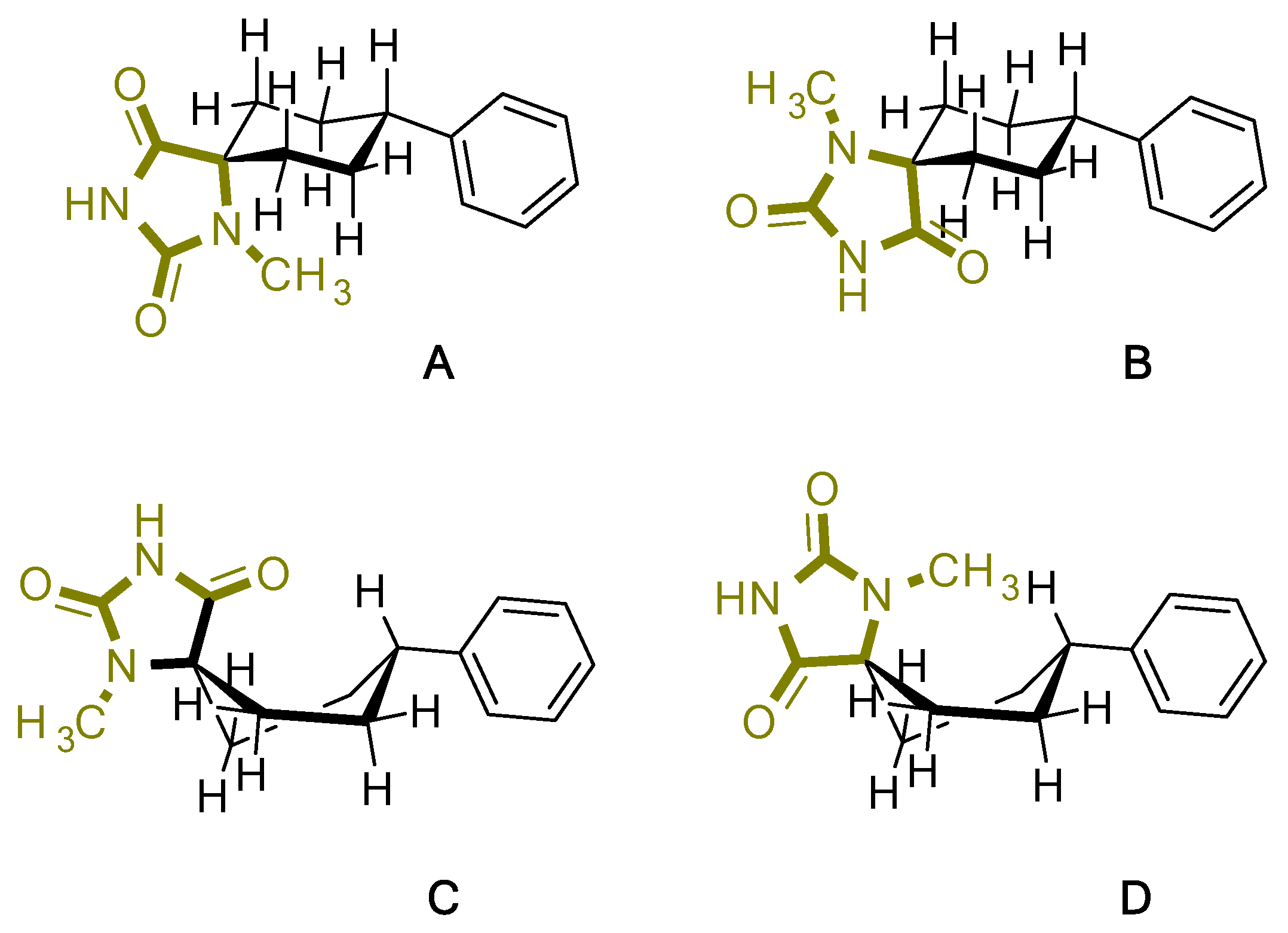
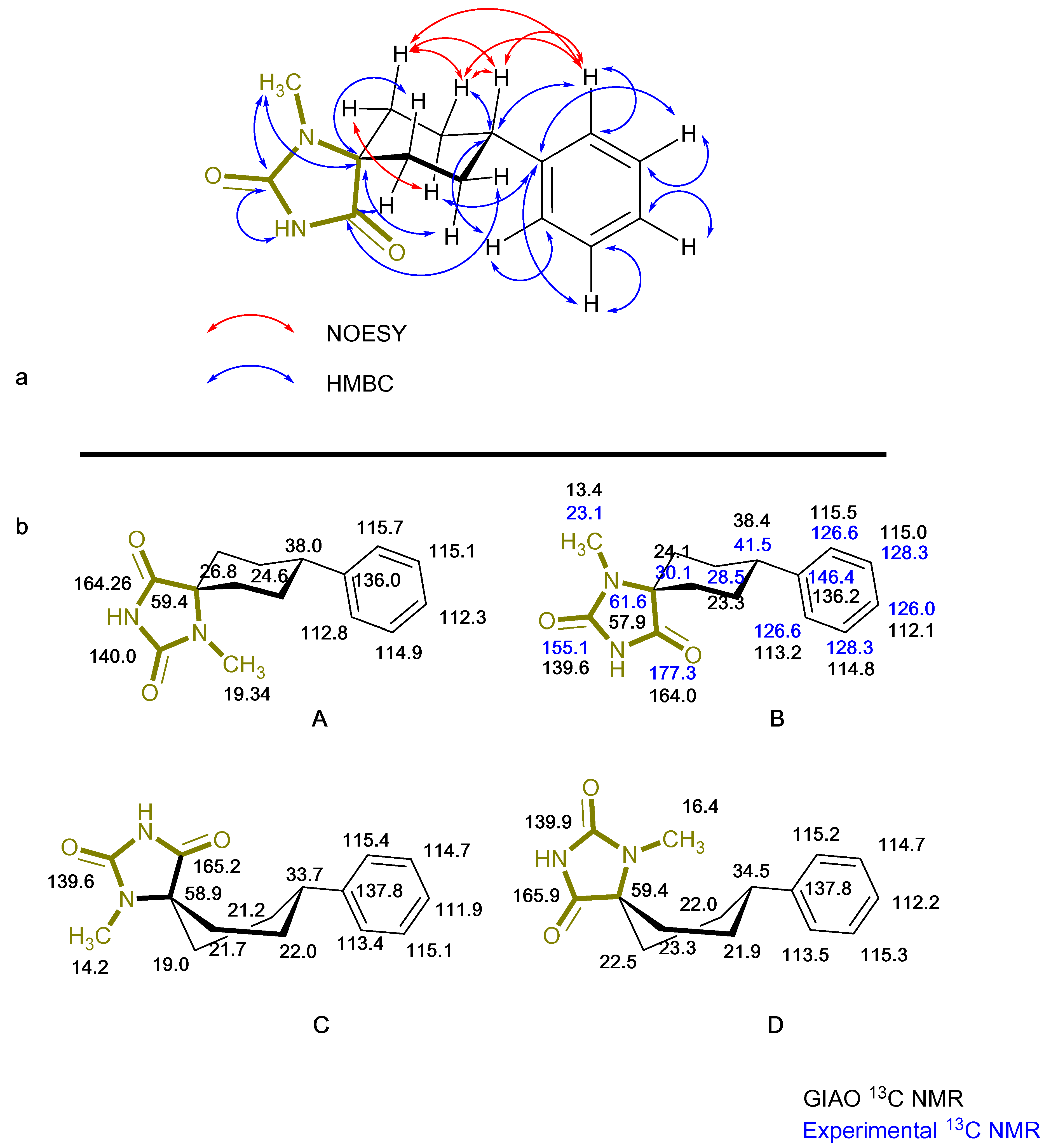
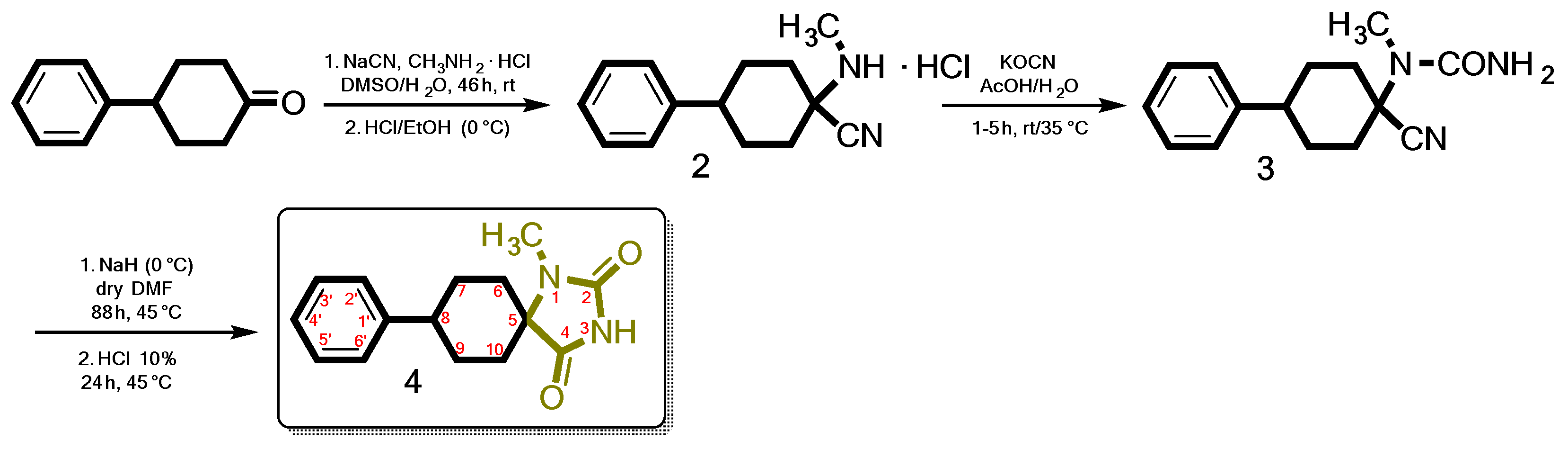
2. Results
3. Materials and Methods
3.1. Chemistry
3.2. Computational
3.3. Synthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cho, S.H.; Kim, S.-H.; Shin, D. Recent applications of hydantoin and thiohydantoin in medicinal chemistry. Eur. J. Med. Chem. 2019, 164, 517–545. [Google Scholar] [CrossRef] [PubMed]
- Trišović, N.; Valentić, N.; Ušćumlić, G. Solvent effects on the structure-property relationship of anticonvulsant hydantoin derivatives: A solvatochromic analysis. Chem. Cent. J. 2011, 5, 62. [Google Scholar] [CrossRef]
- Iqbal, Z.; Ali, S.; Iqbal, J.; Abbas, Q.; Qureshi, I.Z.; Hameed, S. Dual action spirobicycloimidazolidine-2,4-diones: Antidiabetic agents and inhibitors of aldose reductase-an enzyme involved in diabetic complications. Bioorg. Med. Chem. Lett. 2013, 23, 488–491. [Google Scholar] [CrossRef]
- Zhang, M.; Liang, Y.R.; Li, H.; Liu, M.M.; Wang, Y. Design, synthesis, and biological evaluation of hydantoin bridged analogues of combretastatin A-4 as potential anticancer agents. Bioorg. Med. Chem. 2017, 25, 6623–6634. [Google Scholar] [CrossRef]
- Czopek, A.; Byrtus, H.; Zagórska, A.; Siwek, A.; Kazek, G.; Bednarski, M.; Sapa, J.; Pawłowski, M. Design, synthesis, anticonvulsant, and antiarrhythmic properties of novel N-Mannich base and amide derivatives of β-tetralinohydantoin. Pharmacol. Rep. 2016, 68, 886–893. [Google Scholar] [CrossRef]
- Wang, Z.D.; Sheikh, S.O.; Zhang, Y. A Simple Synthesis of 2-Thiohydantoins. Molecules 2006, 11, 739–750. [Google Scholar] [CrossRef]
- Kopsky, D.J.; Keppel Hesselink, J.M. Phenytoin Cream for the Treatment of Neuropathic Pain: Case Series. Pharmaceuticals 2018, 11, 53. [Google Scholar] [CrossRef]
- Hosoya, T.; Aoyama, H.; Ikemoto, T.; Hiramatsu, T.; Kihara, Y.; Endo, M.; Suzuki, M. Dantrolene analogues revisited: General synthesis and specific functions capable of discriminating two kinds of Ca2+ release from sarcoplasmic reticulum of mouse skeletal muscle. Bioorg. Med. Chem. 2003, 11, 663–673. [Google Scholar] [CrossRef]
- Last-Barney, K.; Davidson, W.; Cardozo, M.; Frye, L.L.; Grygon, C.A.; Hopkins, J.L.; Jeanfavre, D.D.; Pav, S.; Stevenson, J.M.; Tong, L.; et al. Binding Site Elucidation of Hydantoin-Based Antagonists of LFA-1 Using Multidisciplinary Technologies: Evidence for the Allosteric Inhibition of a Protein−Protein Interaction. J. Am. Chem. Soc. 2001, 123, 5643–5650. [Google Scholar] [CrossRef]
- Giannakopoulou, E.; Pardali, V.; Skrettas, I.; Zoidis, G. Transesterification instead of N-Alkylation: An Intriguing Reaction. ChemistrySelect 2019, 4, 3195–3198. [Google Scholar] [CrossRef]
- Giannakopoulou, E.; Pardali, V.; Frakolaki, E.; Siozos, V.; Myrianthopoulos, V.; Mikros, E.; Taylor, M.C.; Kelly, J.M.; Vassilaki, N.; Zoidis, G. Scaffold hybridization strategy towards potent hydroxamate-based inhibitors of Flaviviridae viruses and Trypanosoma species. Med. Chem. Commun. 2019, 10, 991–1006. [Google Scholar] [CrossRef]
- Dragojlovic, V. Conformational analysis of cycloalkanes. ChemTexts 2015, 1, 1–30. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li–F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 2002, 112, 8251–8260. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- McWeeny, R. Perturbation Theory for the Fock-Dirac Density Matrix. Phys. Rev. 1962, 126, 1028–1034. [Google Scholar] [CrossRef]
- London, F. Théorie quantique des courants interatomiques dans les combinaisons aromatiques. J. Phys. Radium 1937, 8, 397–409. [Google Scholar] [CrossRef]
- Deng, W.; Cheeseman, J.R.; Frisch, M.J. Calculation of Nuclear Spin-Spin Coupling Constants of Molecules with First and Second Row Atoms in Study of Basis Set Dependence. J. Chem. Theory Comput. 2006, 2, 1028–1037. [Google Scholar] [CrossRef]
- Peralta, J.E.; Scuseria, G.E.; Cheeseman, J.R.; Frisch, M.J. Basis set dependence of NMR spin–spin couplings in density functional theory calculations: First row and hydrogen atoms. Chem. Phys. Lett. 2003, 375, 452–458. [Google Scholar] [CrossRef]
- Barone, V.; Peralta, J.E.; Contreras, R.H.; Snyder, J.P. DFT Calculation of NMR JFF Spin−Spin Coupling Constants in Fluorinated Pyridines. J. Phys. Chem. A 2002, 106, 5607–5612. [Google Scholar] [CrossRef]
- Helgaker, T.; Watson, M.; Handy, N.C. Analytical calculation of nuclear magnetic resonance indirect spin–spin coupling constants at the generalized gradient approximation and hybrid levels of density-functional theory. J. Chem. Phys. 2000, 113, 9402–9409. [Google Scholar] [CrossRef]
- Sychrovský, V.R.; Gräfenstein, J.; Cremer, D. Nuclear magnetic resonance spin–spin coupling constants from coupled perturbed density functional theory. J. Chem. Phys. 2000, 113, 3530–3547. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Conformation A | Conformation Β | Conformation C | Conformation D |
|---|---|---|---|---|
| 4 |  |  |  |  |
| Charge | 0 | 0 | 0 | 0 |
| Spin | Singlet | Singlet | Singlet | Singlet |
| Solvation | None | None | None | None |
| E(RB3LYP) | -842.498395 Hartree | -842.501986 Hartree | -842.492052 Hartree | -842.490242 Hartree |
| RMS Gradient Norm | - | - | - | - |
| Imaginary Freq | - | - | - | - |
| Dipole Moment | 3.364844 | 3.032529 Debye | 3.042446 Debye | 3.364416 Debye |
| Point Group | C1 | C1 | C1 | C1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pardali, V.; Katsamakas, S.; Giannakopoulou, E.; Zoidis, G. 1-Methyl-8-phenyl-1,3-diazaspiro[4.5]decane-2,4-dione. Molbank 2021, 2021, M1228. https://doi.org/10.3390/M1228
Pardali V, Katsamakas S, Giannakopoulou E, Zoidis G. 1-Methyl-8-phenyl-1,3-diazaspiro[4.5]decane-2,4-dione. Molbank. 2021; 2021(2):M1228. https://doi.org/10.3390/M1228
Chicago/Turabian StylePardali, Vasiliki, Sotirios Katsamakas, Erofili Giannakopoulou, and Grigoris Zoidis. 2021. "1-Methyl-8-phenyl-1,3-diazaspiro[4.5]decane-2,4-dione" Molbank 2021, no. 2: M1228. https://doi.org/10.3390/M1228
APA StylePardali, V., Katsamakas, S., Giannakopoulou, E., & Zoidis, G. (2021). 1-Methyl-8-phenyl-1,3-diazaspiro[4.5]decane-2,4-dione. Molbank, 2021(2), M1228. https://doi.org/10.3390/M1228