
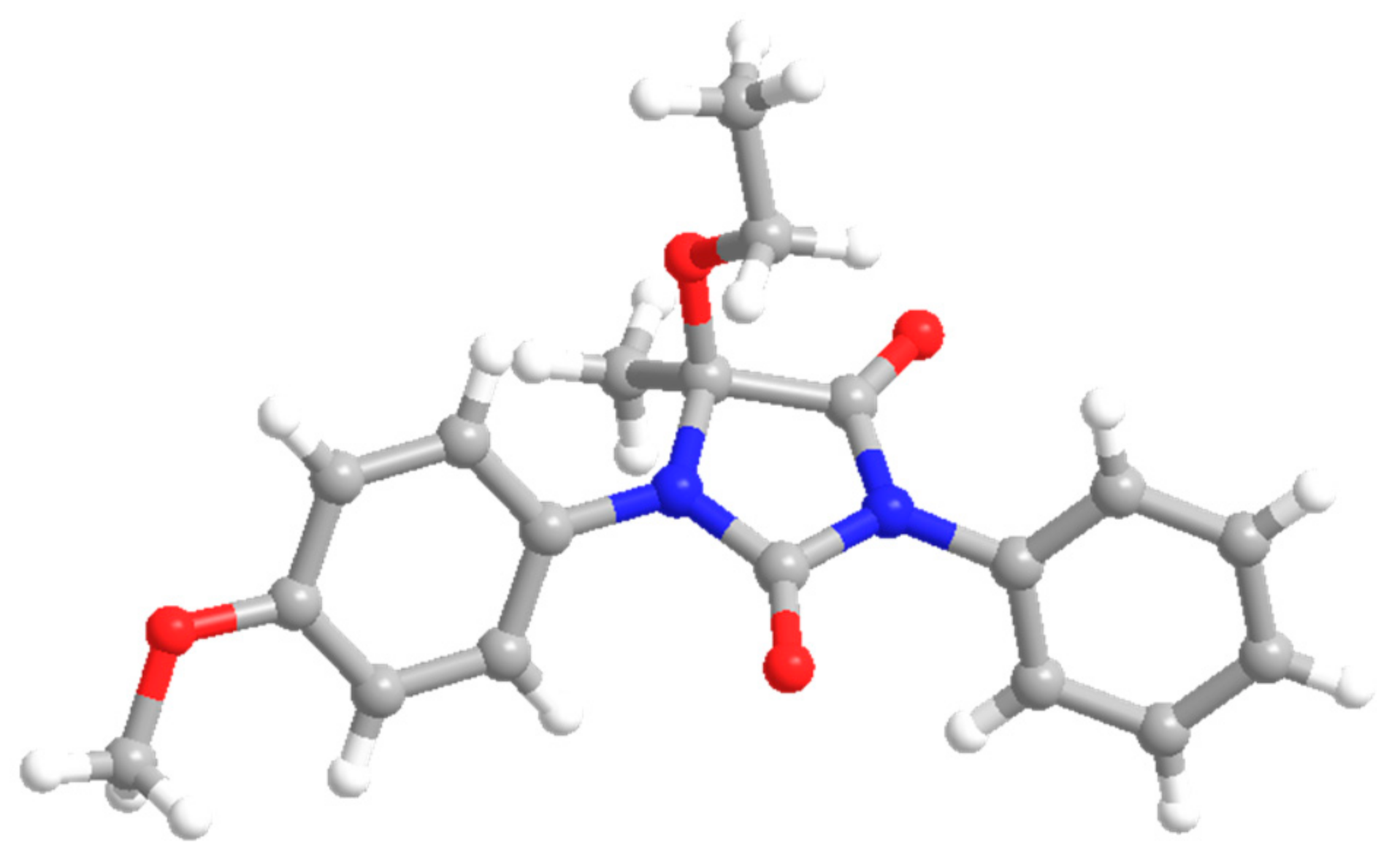
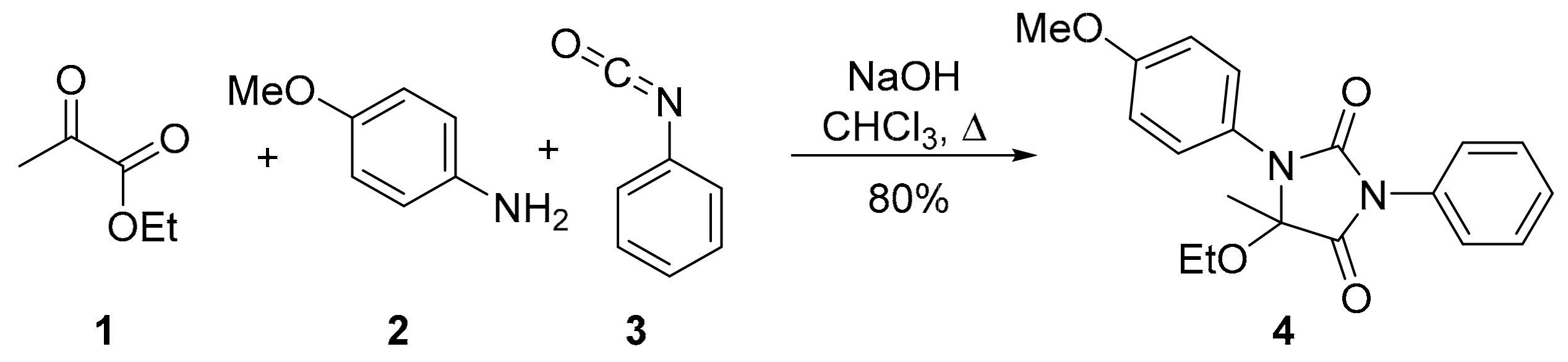
5-Ethoxy-1-(4-methoxyphenyl)-5-methyl-3-phenylimidazolidine-2,4-dione

 ,
,  , ,
, , 

Abstract
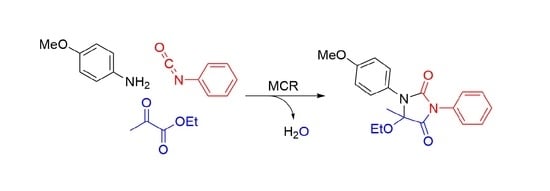
{kind=link}
{kind=link}
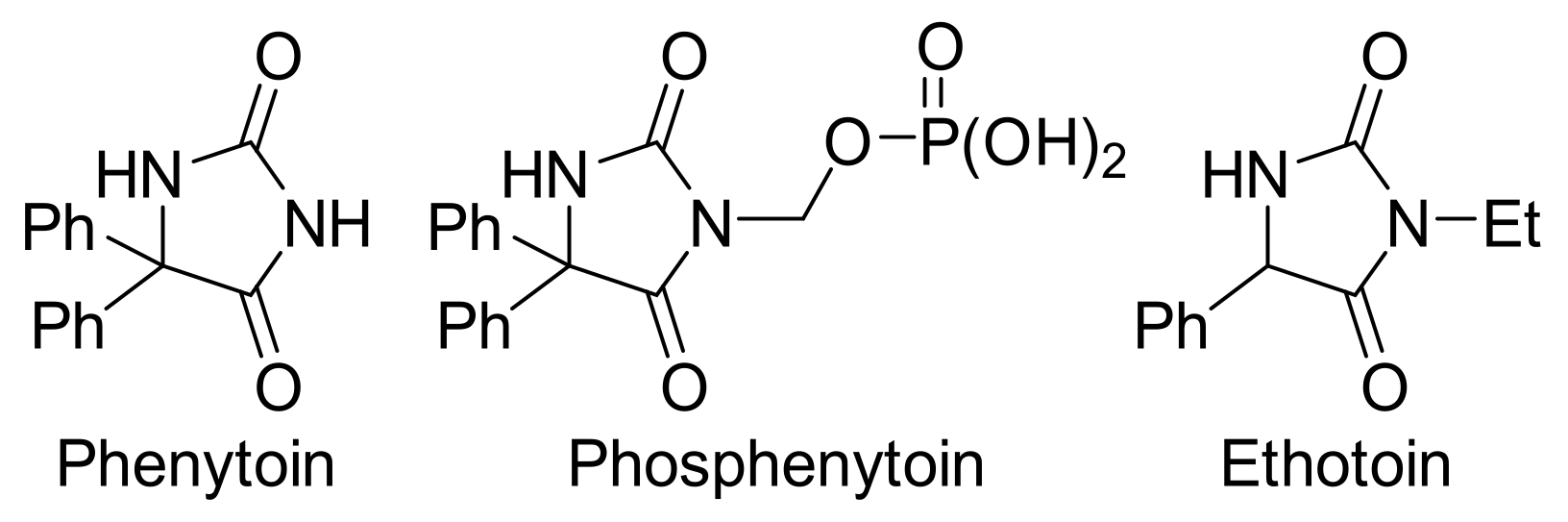
{kind=link}
{kind=link}
{kind=link}
{kind=link}
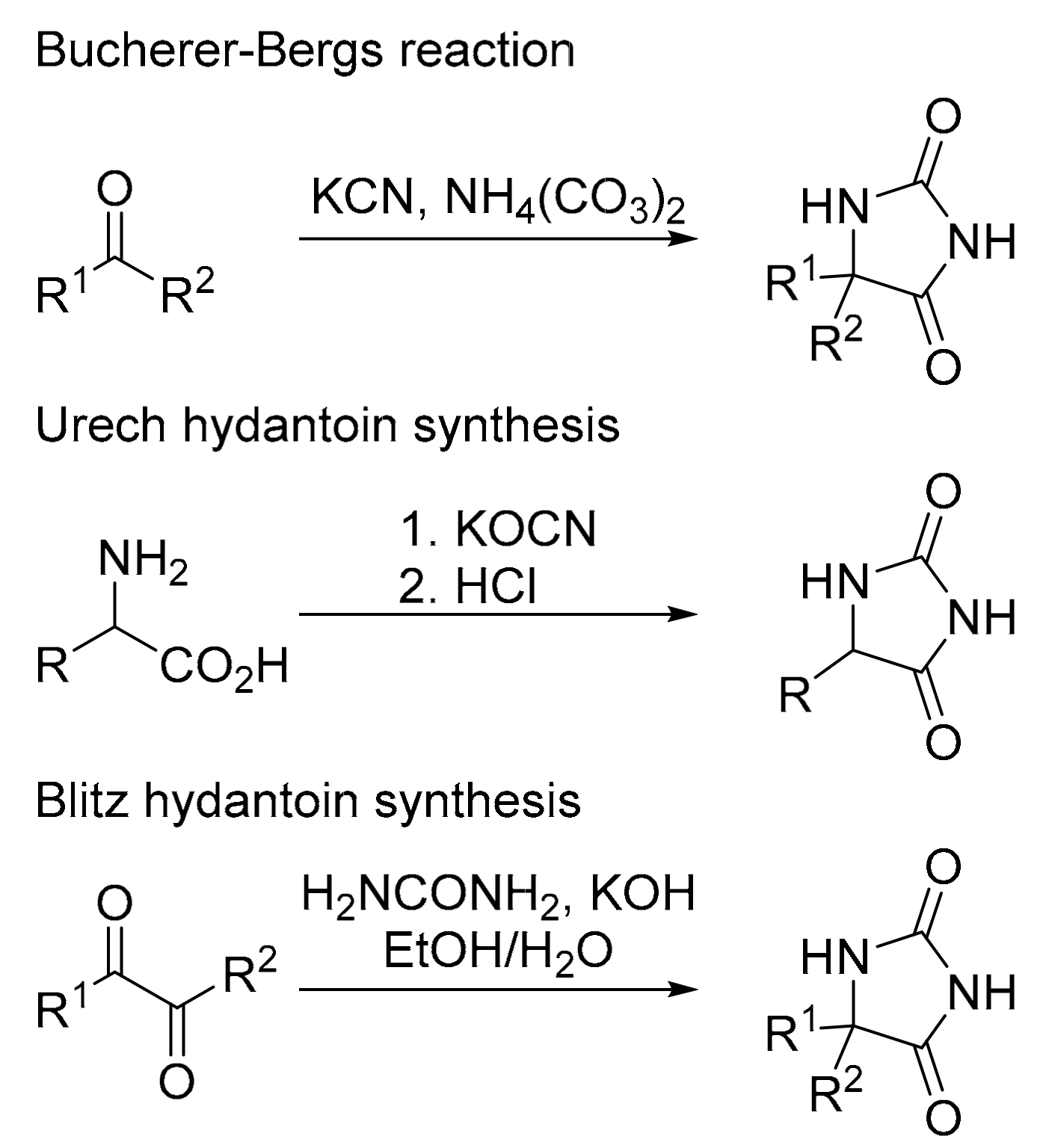
1. Introduction
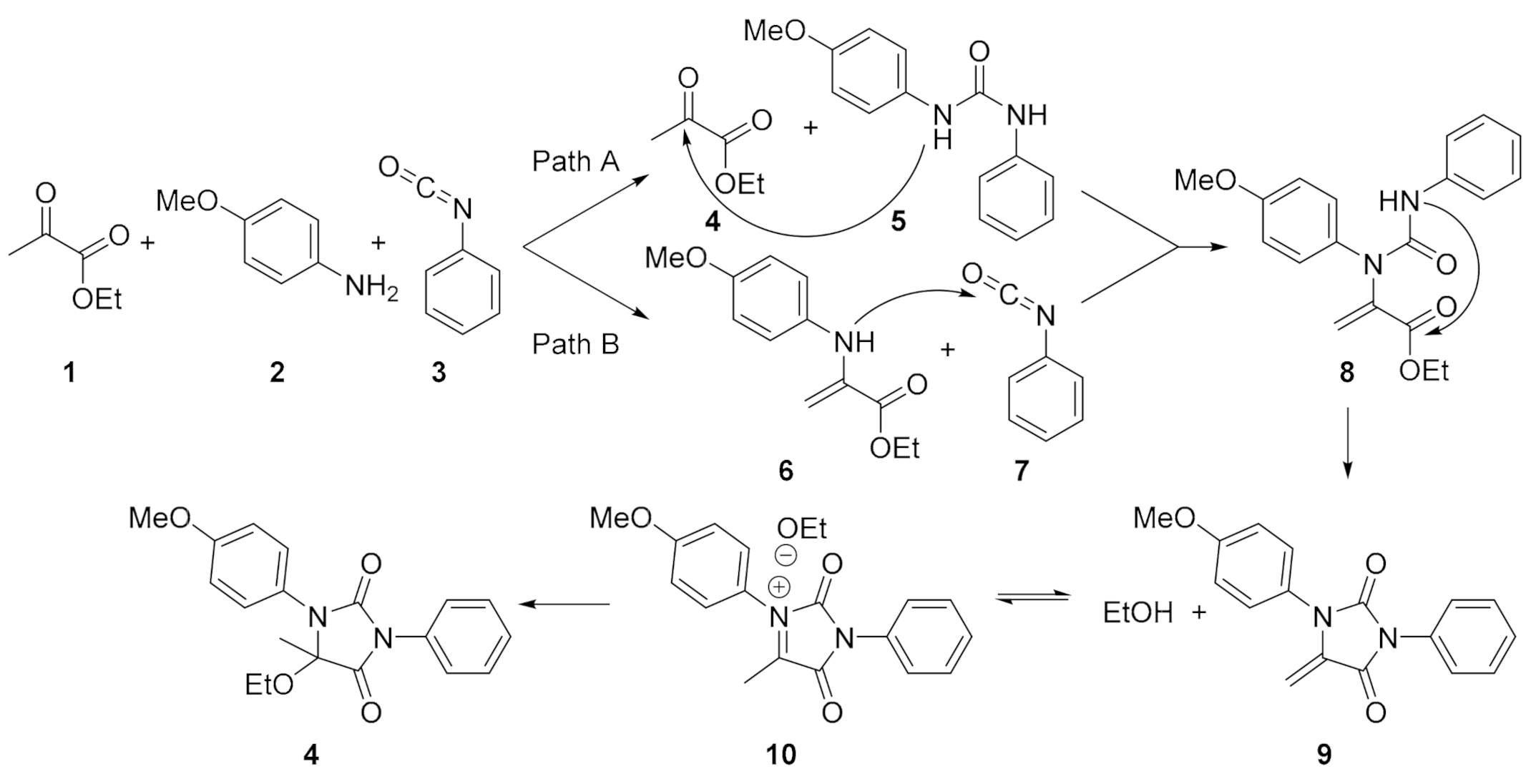
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Information
3.2. Experimental Procedures and Characterization Data for Hydantoin 4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Baeyer, A. Beiträge zur Kenntniss der Harnsäuregruppe. Justus Liebigs Ann. Chem. 1861, 119, 126–128. [Google Scholar] [CrossRef]
- LoVecchio, F. Hydantoin Anticonvulsants: Phenytoin and Fosphenytoin. In Synthesis of Essential Drugs; Brent, J., Burkhart, K., Dargan, P., Hatten, B., Mégarbane, B., Palmer, R., White, J., Eds.; Springer International Publishing: Basel, Switzerland, 2017. [Google Scholar] [CrossRef]
- Brown, M.L.; Brown, G.B.; Brouillette, W.J. Effects of log P and phenyl ring conformation on the binding of 5-phenylhydantoins to the voltage-dependent sodium channel. J. Med. Chem. 1997, 40, 602–607. [Google Scholar] [CrossRef]
- Vardanyan, R.S.; Hruby, V.J. (Eds.) Antiepileptic Drugs. In Critical Care Toxicology; Elsevier: Amsterdam, The Netherlands, 2006; pp. 125–133. [Google Scholar] [CrossRef]
- Volonterio, A.; Ramirez de Arellano, C.; Zanda, M. Synthesis of 1,3,5-Trisubstituted Hydantoins by Regiospecific Domino Condensation/Aza-Michael/O→N Acyl Migration of Carbodiimides with Activated α,β-Unsaturated Carboxylic Acids. J. Org. Chem. 2005, 70, 2161–2170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ye, D.; Feng, E.; Wang, J.; Shi, J.; Jiang, H.; Liu, H. Highly α-Selective Synthesis of Sialyl Spirohydantoins by Regiospecific Domino Condensation/O→N Acyl Migration/N-Sialylation of Carbodiimides with Peracetylated Sialic Acid. J. Org. Chem. 2010, 75, 3552–3557. [Google Scholar] [CrossRef]
- Brouillette, Y.; Lisowski, V.; Guillon, J.; Massip, S.; Martinez, J. Efficient one-pot microwave-assisted synthesis of 3-(thien-3-yl)imidazolidine-2,4-dione analogs. Tetrahedron 2007, 63, 7538–7544. [Google Scholar] [CrossRef]
- Mandal, A.; Krishnan, R.S.G.; Thennarasu, S.; Panigrahi, S.; Mandal, A.B. Two-dimensional surface properties of an antimicrobial hydantoin at the air–water interface: An experimental and theoretical study. Colloids Surf. B 2010, 79, 136–141. [Google Scholar] [CrossRef]
- Todorov, P.T.; Naydenova, E.D. Synthesis and characterization of novel dipeptide mimetics with hydantoin moiety. C. R. Chim. 2010, 13, 1424–1428. [Google Scholar] [CrossRef]
- Kumar, C.A.; Swamy, S.N.; Sugahara, K.; Rangappa, K.S. Anti-tumor and anti-angiogenic activity of novel hydantoin derivatives: Inhibition of VEGF secretion in liver metastatic osteosarcoma cells. Bioorg. Med. Chem. 2009, 17, 4928–4934. [Google Scholar] [CrossRef]
- Gong, Y.D.; Sohn, H.Y.; Kurth, M.J. Microwave-Mediated Intramolecular Carbanilide Cyclization to Hydantoins Employing Barium Hydroxide Catalysi. J. Org. Chem. 1998, 63, 4854–4856. [Google Scholar] [CrossRef]
- Alizadeh, A.; Sheikhi, E. One-pot synthesis of functionalized hydantoin derivatives via a four-component reaction between an amine, an arylsulfonyl isocyanate and an alkyl propiolate or dialkyl acetylenedicarboxylate in the presence of triphenylphosphine. Tetrahedron Lett. 2007, 48, 4887–4890. [Google Scholar] [CrossRef]
- Marton, J.; Enisz, J.; Hosztafi, S.; Timar, T. Preparation and fungicidal activity of 5-substituted hydantoins and their 2-thio analogs. J. Agric. Food Chem. 1993, 41, 148–152. [Google Scholar] [CrossRef]
- Bucherer, H.T.; Lieb, V.A. About the formation of substituted hydantoine from aldehydes and ketones—Synthesis of hydantoin (II announcement). J. Prakt. Chem. 1934, 141, 5–43. [Google Scholar] [CrossRef]
- Urech, F. XXI. Ueber Lacturaminsäure und Lactylharnstoff. Liebigs Ann. 1873, 165, 99–103. [Google Scholar] [CrossRef]
- Biltz, H. Über die Konstitution der Einwirkungsprodukte von substituierten Harnstoffen auf Benzil und über einige neue Methoden zur Darstellung der 5.5-Diphenyl-hydantoine. Ber. Dtsch. Chem. Ges. 1908, 41, 1379–1393. [Google Scholar] [CrossRef]
- Ware, E. The Chemistry of the Hydantoins. Chem. Rev. 1950, 46, 403–470. [Google Scholar] [CrossRef] [PubMed]
- López, C.A.; Trigo, G.G. The Chemistry of Hydantoins. Adv. Heterocycl. Chem. 1985, 38, 177–228. [Google Scholar] [CrossRef]
- Meusel, M.; Gütschow, M. Recent Developments in Hydantoin Chemistry. A Review. Org. Prep. Proced. Int. 2004, 36, 391–443. [Google Scholar] [CrossRef]
- Konnert, L.; Lamaty, F.; Martínez, J.; Colaciano, E. Recent Advances in the Synthesis of Hydantoins: The State of the Art of a Valuable Scaffold. Chem. Rev. 2017, 117, 13757–13809. [Google Scholar] [CrossRef]
- Hulme, C.; Bienaymé, H.; Nixey, T.; Chenera, B.; Jones, W.; Tempest, P.; Smith, A.L. Library Generation via Postcondensation Modifications of Isocyanide-Based Multicomponent Reactions. Methods Enzym. 2003, 369, 469–496. [Google Scholar] [CrossRef]
- Cioc, R.C.; Ruijter, E.; Orru, R.V.A. Multicomponent reactions: Advanced tools for sustainable organic synthesis. Green Chem. 2014, 16, 2958–2975. [Google Scholar] [CrossRef]
- Kotha, S.; Gupta, N.K.; Aswar, V.R. Multicomponent Approach to Hydantoins and Thiohydantoins Involving a Deep Eutectic Solvent. Chem. Asian, J. 2019, 14, 3188–3197. [Google Scholar] [CrossRef]
- Slobbe, P.; Ruijter, E.; Orru, R.V.A. Recent applications of multicomponent reactions in medicinal chemistry. Med. Chem. Commun. 2012, 3, 1189–1218. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, Q.; Wang, M.-X. Multicomponent Reactions in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Müller, T.J.J. Science of Synthesis, Multicomponent Reactions, Vol 1 and 2; Thieme: Stutgart, Germany, 2014. [Google Scholar]
- Palacios, F.; Vicario, J.; Aparicio, D. An efficient synthesis of achiral and chiral cyclic dehydro-α-amino acid derivatives through nucleophilic addition of Amines to β,γ-unsaturated α-keto esters. Eur. J. Org. Chem. 2006, 2006, 2843–2850. [Google Scholar] [CrossRef]
- del Corte, X.; Maestro, A.; Vicario, J.; Martínez de Marigorta, E.; Palacios, F. Brönsted-Acid-Catalyzed Asymmetric Three-Component Reaction of Amines, Aldehydes, and Pyruvate Derivatives. Enantioselective Synthesis of Highly Functionalized γ-Lactam Derivatives. Org. Lett. 2018, 20, 317–320. [Google Scholar] [CrossRef] [PubMed]
- del Corte, X.; Martínez de Marigorta, E.; Palacios, F.; Vicario, J. A Brønsted Acid-Catalyzed Multicomponent Reaction for the Synthesis of Highly Functionalized γ-Lactam Derivatives. Molecules 2019, 24, 2951. [Google Scholar] [CrossRef]
- del Corte, X.; López-Francés, A.; Maestro, A. Martínez de Marigorta, E.; Palacios, F.; Vicario. J. Brönsted Acid Catalyzed Multicomponent Synthesis of Phosphorus and Fluorine-Derived γ-Lactam Derivatives. J. Org. Chem. 2020, 85, 14369–14383. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
del Corte, X.; López-Francés, A.; Martinez de Marigorta, E.; Palacios, F.; Vicario, J. 5-Ethoxy-1-(4-methoxyphenyl)-5-methyl-3-phenylimidazolidine-2,4-dione. Molbank 2021, 2021, M1218. https://doi.org/10.3390/M1218
del Corte X, López-Francés A, Martinez de Marigorta E, Palacios F, Vicario J. 5-Ethoxy-1-(4-methoxyphenyl)-5-methyl-3-phenylimidazolidine-2,4-dione. Molbank. 2021; 2021(2):M1218. https://doi.org/10.3390/M1218
Chicago/Turabian Styledel Corte, Xabier, Adrián López-Francés, Edorta Martinez de Marigorta, Francisco Palacios, and Javier Vicario. 2021. "5-Ethoxy-1-(4-methoxyphenyl)-5-methyl-3-phenylimidazolidine-2,4-dione" Molbank 2021, no. 2: M1218. https://doi.org/10.3390/M1218
APA Styledel Corte, X., López-Francés, A., Martinez de Marigorta, E., Palacios, F., & Vicario, J. (2021). 5-Ethoxy-1-(4-methoxyphenyl)-5-methyl-3-phenylimidazolidine-2,4-dione. Molbank, 2021(2), M1218. https://doi.org/10.3390/M1218