1. Introduction
Plant-derived compounds have successfully inspired the drug discovery process which has exploited the entire structure of such natural products or part of that to develop new medicinal drugs [
1,
2].
Polyphenols and related compounds are secondary metabolites which occur naturally in plants and which have been reported to display protective effects in plant-based diet [
3].
Indeed, food containing polyphenols are known to exert beneficial effects after long term consumption offering protection against the development of cancers, cardiovascular diseases, diabetes, osteoporosis, and neurodegenerative diseases [
4,
5,
6]. In particular, for some substances such as flavan-3-ols, the beneficial effects are not due to the compounds present
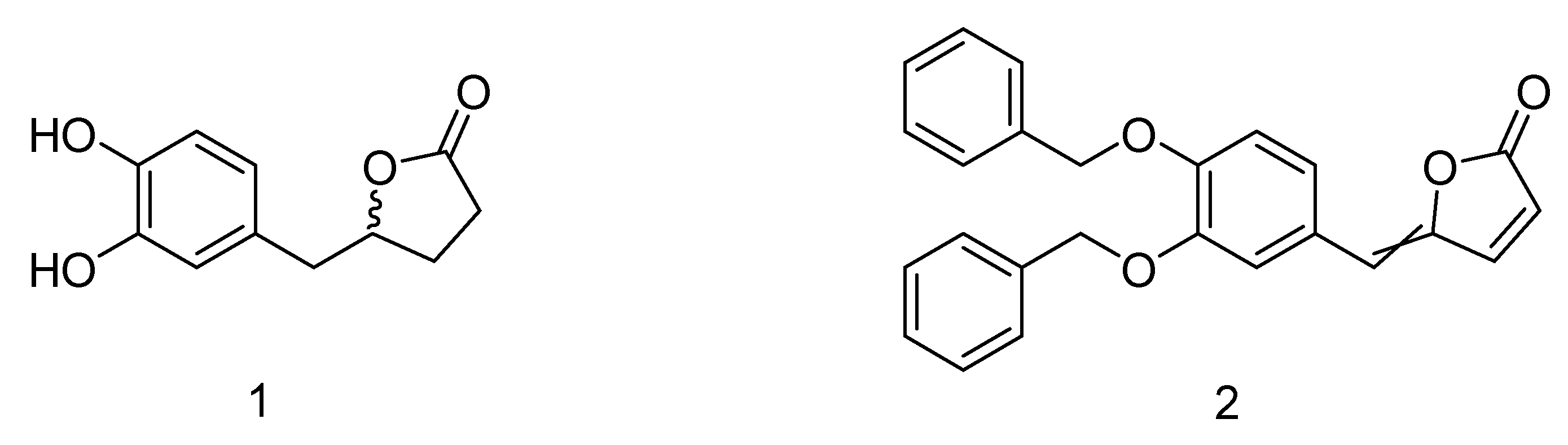
in planta but rather to the related enteric metabolites. As recently reported in the literature by our group, 5-(3′,4′-dihydroxyphenyl)-γ-valerolactone (
1), which was identified as a metabolite in human urine after consumption of standardized and rich polyphenols dietary supplement [
7], displays inhibition of
Candida albicans adhesion.
The synthetic precursor of the 5-(3′,4′-dihydroxyphenyl)-γ-valerolactone (
1) is the γ-alkylidenebutenolide (
2) featured by a α,β-unsaturated γ-lactone core which bears a substituent at the gamma position (
Figure 1).
This structural moiety characterizes in turn a large class of bioactive natural products endowed by a wide range of biological activities, such as inhibition of cholesterol biosynthesis−xerulin (
3a), xerulinic acid (
3b) and diihydroxerulin (
4)—[
8], cytotoxicity−goniobutenolides A (
5a) and B (
5b) and nostoclides (
6)—[
9], antimicrobial activity−rubrolide (
7)—[
10]. Some of these representative γ-alkylidenebutenolide are reported (
Figure 2).
For these reasons stereoselective preparation of γ-alkylidenebutenolide continues to be an important issue for organic chemists.
A considerable amount of naturally occurring γ-alkylidenebutenolide have the Z configuration and most of them lack a β-substituent as demonstrated by compounds 3a, 3b, 4, 5a, and 5b.
Although the
E-isomers are less stable than the
Z-counterparts, in the absence of a β-substituent the formation of
Z-isomers remains small [
11]. Therefore, traditional synthetic strategies including alkylidenation of preformed oxacycles as butanolides and 2-silyloxyfurans, to the best of our knowledge, are nonstereoselective [
12]. Alternative methods offer stereochemical predictability and high level of selectivity but need stereodefined precursors, chiral ligands, or removable stereocontrol elements [
13,
14,
15]. Since the biological activity of compounds featuring a γ-alkylidenebutenolide is due to this core, we focus our attention on preparing the 5-(3′,4′-bis(benzyloxy)benzylidene)furan-2(5
H)-one via a stereocontrolled reaction (
2a Z-isomer,
2b E-isomer;
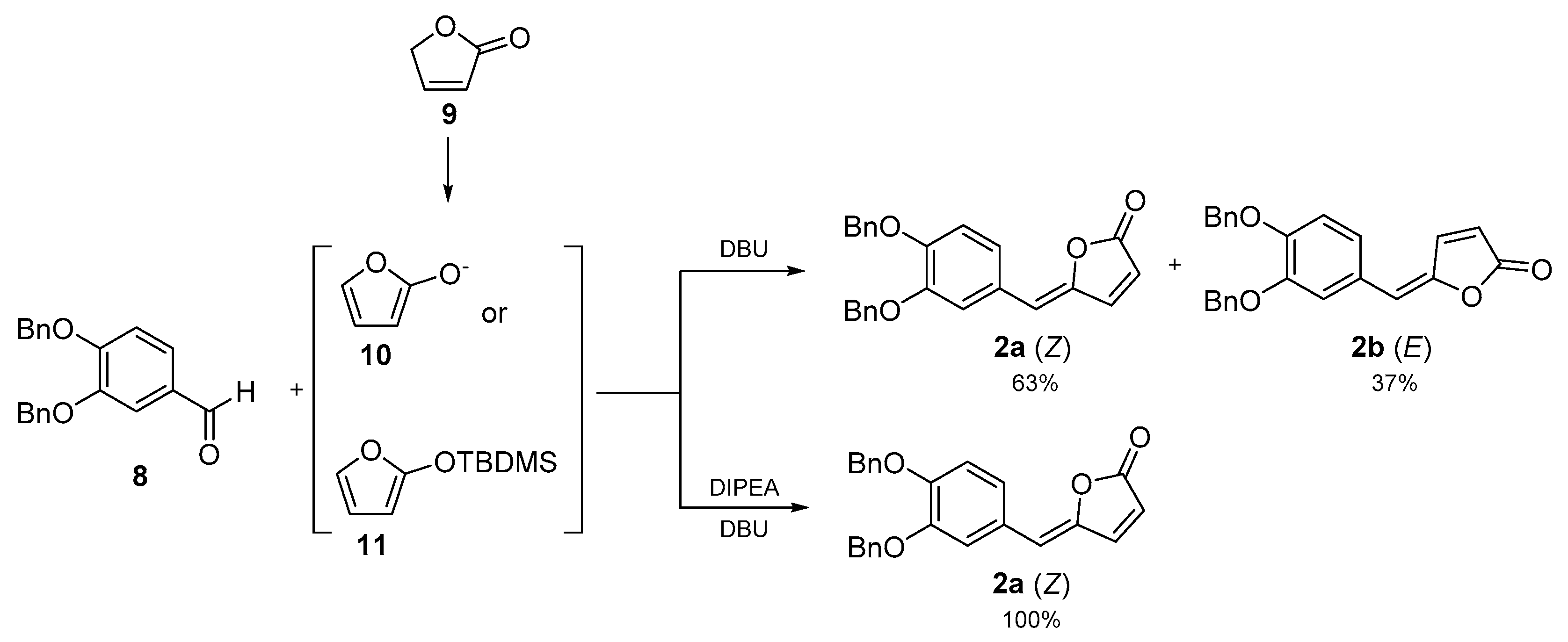
Scheme 1).
2. Results
(
Z)-5-(3′,4′-Bis(benzyloxy)benzylidene)furan-2(5
H)-one
2a was synthesized via vinylogous Mukayama aldol reaction (
Scheme 1). The reaction was performed one-pot by adding diisopropylethylamine (DIPEA) under nitrogen atmosphere to a solution of 2(5
H)-furanone in dry tetrahydrofuran (THF). The mixture was stirred for 30 min at room temperature, and after cooling down to −10 °C,
tert-butyldimethylsilyltrifluoromethanesulfonate and 3,4-bis(benzyloxy)benzaldehyde were added dropwise. The mixture was stirred 2 h and subsequently 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was added. The reaction mixture was stirred overnight at room temperature, then the solvent was removed under vacuum. Optimization of work-up and purification methods gave the title compound as an orange oil (
Z)-isomer (52% yield).
3. Discussion
The vinylogous Mukayama aldol reaction [
16] is the critical step since it drives the stereoselectivity of the γ-alkylidenebutenolide of our interest that is the 5-(3′,4′-bis(benzyloxy)benzylidene)furan-2(5
H)-one.
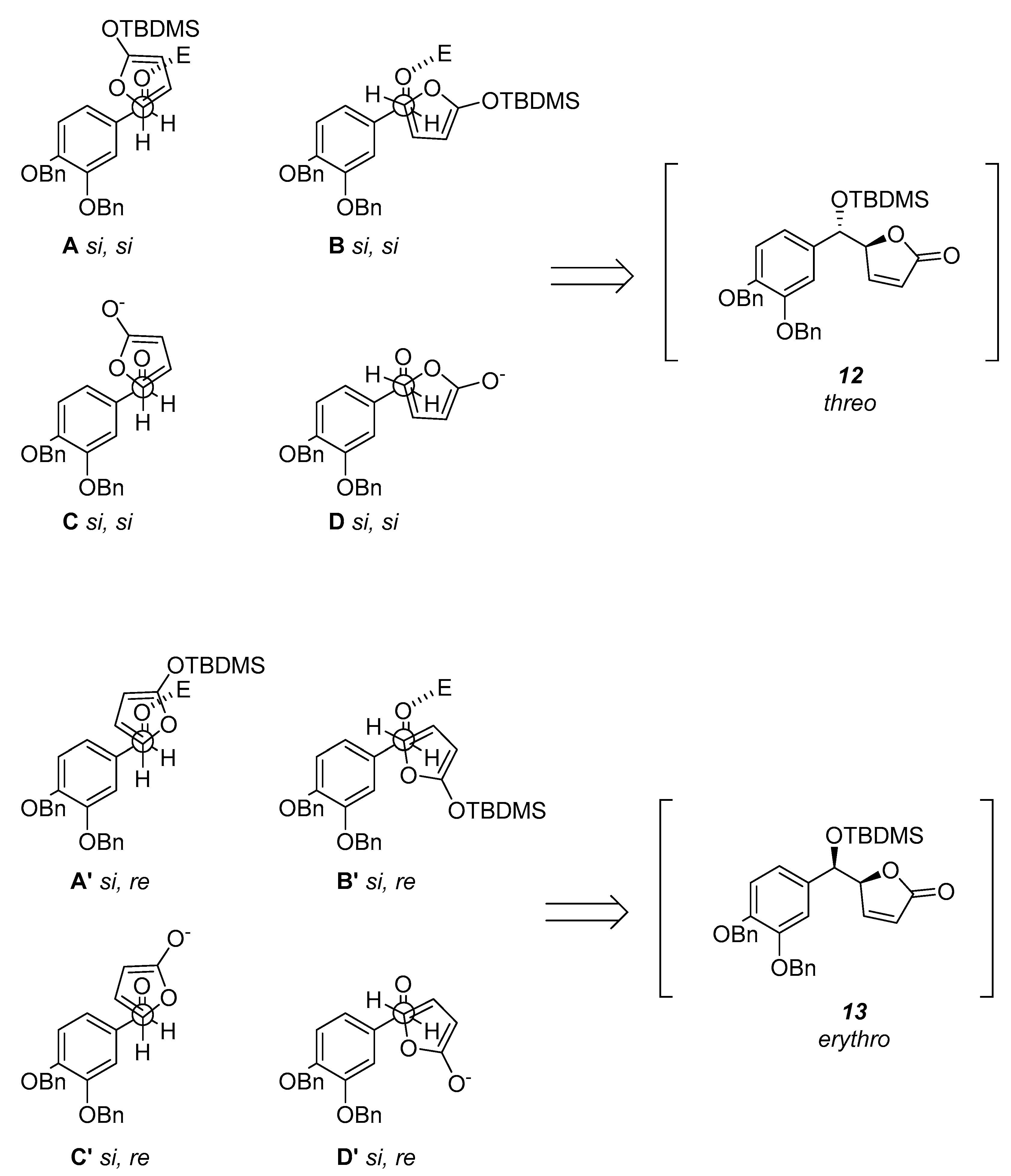
We decided to use tert-butyldimethylsilyltrifluoromethanesulfonate (TBDMSOTf) which serves as both silylating agent and Lewis acid catalyst. We began our investigation treating the commercially available 2(5H)-furanone with DBU either to form the intermediate 2-tert-butyldimethylsiloxyfuran (11) or to dehydrate it. After several unsatisfactory attempts in which we obtained the mixture of the E- and Z-isomers we started an insightful theoretical investigation of the possible transition states.
Encounter between the C1 and C5 atoms of the aldehyde and furan respectively could occur through the
si faces or the
si and
re faces. Accordingly to what was reported by Jefford et al. [
17], the resultant two pairs of staggered arrangements
A,
B and,
C,
D will produce the
threo (
12), while the other two pairs
A’,
B’ and,
C’,
D’ will lead to the formation of the
erythro (
13) aldols (
Scheme 2). Moreover, in accordance with the vinylogous Mukayama reaction mechanism the Lewis acid TBDMSOTf coordinates the aldehyde with
syn orientation. Thus, the prevalence of one arrangement over the other depends only on steric and electronic factors. The least hindered arrangements should be
A and
B’. Since the arrangement
A is Diels–Alder like it is favored and it pushes for the
threo selectivity.
The anion equivalent 10 would avoid the arrangements C and C’ because the charge would be better dispersed in D and D’ as confirmed by the density-functional theory (DFT) calculations (D’ = 0.00 kcal/mol; D = +2.11 kcal/mol; C’ = +6.99 kcal/mol; C = +4.44 kcal/mol).
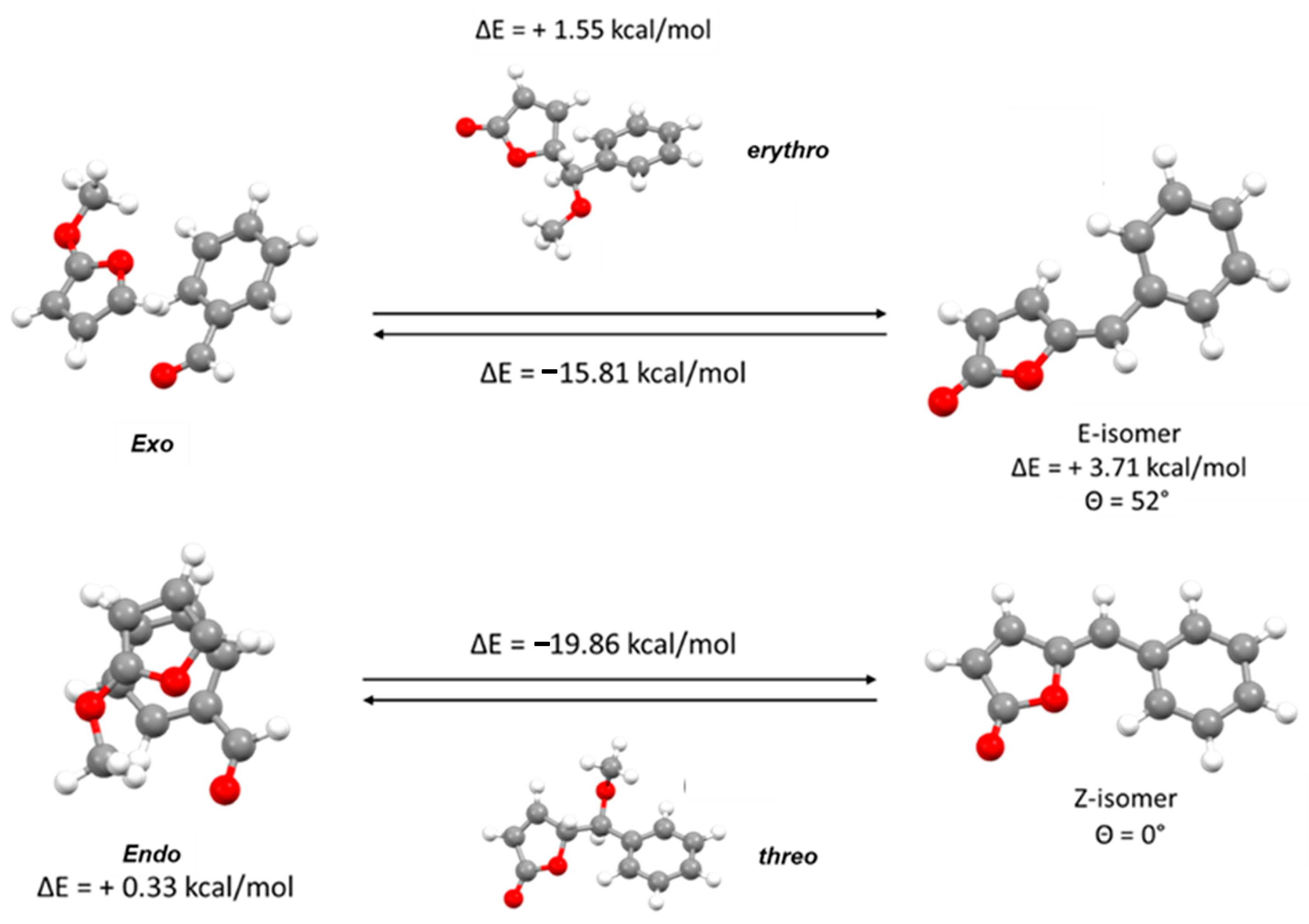
We also applied the DFT quantum mechanical calculations to further investigate the Diels–Alder-like arrangements. To simplify the calculation and to focus attention on the simulated vinylogous Mukayama aldol reaction, the tert-butyldimethylsilyl group was replaced by a methoxy group, while the benzyloxy substituents were removed.
As shown in
Figure 3, the DFT quantum mechanical calculations allow to reach five main considerations:
The exo-conformation is slightly favored compared to the endo-conformation, although this result could be influenced by the lesser steric hindrance of the methoxy group, simulated instead of the TBDMS group for simplicity;
The vinylogous Mukayama aldol reaction is energetically possible, whether the intermediate assume exo or endo-conformation;
The product obtained from endo-conformation is however clearly favored and this trend is already observed in the chiral intermediate. This result is very evident both in the different stability of the two final products and consequently in the different energy profile of the two reactions;
Focusing on the two final products, the higher stability of the Z-isomer is clearly because only this isomer is able to assume a completely planar conformation that allows a full delocalization between the two rings.
- 5.
Moreover, a weak non-classical intramolecular hydrogen bond involving C’6-H’6-O1 shown in
Figure 4 plays an important role in the stabilization of the
Z-isomer.
Considering these assumptions, we used DIPEA for the formation of the 2-tert-butyldimethylsiloxyfuran to enhance a diastereoselectivity coming from the Diels–Alder transition state since the use of the DBU alone allows the formation of anion 10 which would adopt D and D’ arrangements giving reason to the E/Z mixture formation thus a partial threo diastereoselectivity. As supposed and well explained by the Diels–Alder-like orientation of the reactants and by the quantum mechanics calculations, the use of DIPEA for the formation of 12 and DBU to eliminate the TMDMOH gave only the Z-isomer.
The obtained compound exhibited the expected spectroscopic signals (
1H-NMR,
13C-NMR, ESI-MS and HRMS in the
Supplementary Materials) confirming the successful synthesis of
Z-γ-alkylidenebutenolide
2a. Using
Figure 4 as reference, the
1H-NMR (CDCl
3) spectrum shows confirmatory assignments accordingly to Chang et al. [
18] such as the doublet and the singlet integrating as one hydrogen each for H3 and H6 (6.15 d,
J = 5.3 Hz and 5.91 s, 1H).
4. Materials and Methods
2(5H)-Furanone, tert-butyldimethylsilyltrifluoromethanesulfonate (TBDMSOTf), 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), sodium bisulfite, 37% HCl, cyclohexane, ethyl acetate, and tetrahydrofuran were purchased from Merck KGaA, Darmstadt, Germany. 1H-NMR spectra were recorded using a FT-spectrometer operating at 300 MHz while 13C-NMR at 75.43 MHz. Chemical shifts are reported in ppm relative to the residual solvent (CHCl3, MeOH, or DMSO) as an internal standard. Signal multiplicity is designed according to the following abbreviations: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, m = multiplet, br s = broad singlet, br t = broad triplet. Purifications were performed by flash chromatography using silica gel (particle size 40–63 μm, Merck) on IsoleraTM (Biotage, Uppsala, Sweden) apparatus.
The reaction paths were monitored by simulating the involved reactants and products at their ground-state and at gas phase by density functional theory (DFT) using the Becke three-parameter hybrid function with LYP correlation (DFT/B3LYP) and with the 6-31G basis set as implemented by the GAMESS software [
19].
The MS analysis was performed on a TSQ Quantum Ultra Triple Quadrupole (ThermoFinnigan Italy, Milan, Italy) equipped with an ESI source. The following source parameters were used: positive ion mode, capillary temperature 275 °C, spray voltage 4 kV, capillary voltage 33 V, tube lens voltage 120 V. The flow rate of the nebulizer gas (nitrogen) was 10 a.u. The mass spectrometer acquired in full mass scan and the Q3 was used as detector with a scan range 200–700 m/z. The HRMS analysis was performed on a LTQ Orbitrap XL (Thermo Scientific, Bremen, Germany) equipped with an ESI source. The following source parameters were used: positive ion mode, capillary temperature 275 °C, spray voltage 4 kV, capillary voltage 30 V, tube lens offset 90 V. The flow rate of the nebulizer gas (ni-trogen) was 10 a.u. The mass spectrometer acquired in full mass scan, with a scan range of 200–600 m/z and resolution 30,000 FWHM at m/z 400.
4.1. 5-(3′,4′-Bis(benzyloxy)benzylidene)furan-2(5H)-one (2)
4.1.1. (Z)-Isomer Procedure
DIPEA (0.27 mL, 1.53 mmol) was added under nitrogen atmosphere to a solution of 2(5H)-furanone (42.88 mg, 0.51 mmol) in dry THF (3.5 mL). The mixture was stirred for 30 min at room temperature, and after cooling down to −10 °C, tert-butyldimethylsilyltrifluoromethanesulfonate (0.13 mL, 0.56 mmol) and 3,4-bis(benzyloxy)benzaldehyde (141.0 mg, 0.56 mmol) were added dropwise. The mixture was stirred for 2 h at the same temperature and subsequently 2 equivalents of DBU (0.15 mL, 1.02 mmol) were added. The reaction mixture was stirred overnight at room temperature, then solvent was removed under vacuum. Ethyl acetate (20 mL), EtOH (5 mL) and saturated solution of sodium bisulfite (5 mL) were added to the crude residue and stirred overnight at 40 °C. The phases were separated, and the organic layer was diluted with ethyl acetate (15 mL), treated with 2.9 N HCl (3 × 20 mL), washed with brine (20 mL), dried and concentrated to afford a sticky black oil. The crude product was purified on silica gel (75:25 cyclohexane/ethyl acetate) to afford the title compound as an orange oil (100.0 mg, 0.26 mmol, 52% yield). 1H-NMR (300 MHz, CDCl3) Z-isomer: δ = 7.53 (d, J = 7.0 Hz, 4H), 7.49–7.24 (m, 9H), 6.92 (d, J = 8.5 Hz, 1H), 6.15 (d, J = 5.3 Hz, 1H), 5.91 (s, 1H), 5.22 (s, 2H), 5.20 (s, 2H). 13C-NMR (75 MHz, CDCl3): 170.40, 150.15, 148.78, 147.24, 145.16, 145.07, 136.91, 136.75, 128.56 (2), 127.94, 127.67, 127.20, 126.39, 125.33, 125.24, 116.94, 116.46, 116.40, 114.31, 114.27, 114.22, 114.14, 71.23, 70.87. ESI-MS (m/z): calcd. for C25H20O4 384.14, found 385.15 m/z [M + H]+. HRMS calculated [M + H]+ 385.14397 m/z, experimental [M + H]+ 385.14401 m/z (delta ppm: 0.104).
4.1.2. Mixture (Z)- and (E)-Isomers Procedure
DBU (0.28 mL, 1.88 mmol) was added under nitrogen atmosphere to a solution of 2(5H)-furanone (158.0 mg, 1.88 mmol) in dry THF (16 mL). The mixture was stirred for 30 min at room temperature, and after cooling down to −10 °C, tert-butyldimethylsilyl trifluoromethanesulfonate (0.48 mL, 2.07 mmol) and 3,4-bis(benzyloxy)benzaldehyde (600.0 mg, 1.88 mmol) were added dropwise. The mixture was stirred for 1 h at the same temperature and subsequently 2 equivalents of DBU (0.56 mL, 3.77 mmol) were added. The reaction mixture was stirred overnight at room temperature, then solvent was removed under vacuum. Ethyl acetate (20 mL), EtOH (5 mL) and saturated solution of sodium bisulfite (5 mL) were added to the crude residue and stirred overnight at 40 °C. The phases were separated, and the organic layer was diluted with ethyl acetate (15 mL), treated with 2.9 N HCl (3 × 20 mL), washed with brine (20 mL), dried and concentrated to afford a sticky black oil. The crude product was purified on silica gel (75:25 cyclohexane/ethyl acetate) to afford the title compound as an orange/brown oil as a 63:37 mixture of (Z)- and (E)-isomers (354.0 mg, 0.92 mmol, 49% yield). 1H-NMR (300 MHz, CDCl3) Z-isomer: δ = 7.53 (d, J = 7.0 Hz, 4H), 7.49–7.24 (m, 9H), 6.92 (d, J = 8.5 Hz, 1H), 6.15 (d, J = 5.3 Hz, 1H), 5.91 (s, 1H), 5.22 (s, 2H), 5.20 (s, 2H). 1H-NMR (300 MHz, CDCl3) E-isomer: δ = 7.53 (d, J = 7.0 Hz, 4H), 7.49–7.24 (m, 9H), 6.92 (d, J = 8.4 Hz, 1H), 6.21 (d, J = 5.1 Hz, 1H), 5.91 (s, 1H), 5.18 (s, 2H), 5.14 (s, 2H).
Supplementary Materials
The following are available, Page S2: 1H-NMR spectrum of compound 2a, Page S3: 13C-NMR spectrum of compound 2a, Page S4: COSY spectrum of compound 2a, Page S5: ESI-MS spectrum of compound 2a, Page S6: HRMS spectrum of compound 2a, Page S7: 1H-NMR spectrum of compounds 2a/b.
Author Contributions
Conceptualization, L.F.; methodology, L.F. and A.A.; software, G.V.; formal analysis, A.A. and G.B.; investigation, A.A. and G.V.; data curation, A.A. and L.F.; writing—original draft preparation, L.F.; writing—review and editing, L.F. and G.A.; supervision, L.F.; project administration, L.F.; funding acquisition, G.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. The APC was funded by MDPI.
Data Availability Statement
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not available.
References
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural products for drug discovery in the 21st century: Innovations for novel drug discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef] [PubMed]
- Holzmeyer, L.; Hartig, A.K.; Franke, K.; Brandt, W.; Muellner-Riehl, A.N.; Wessjohann, L.A.; Schnitzler, J. Evaluation of plant sources for antiinfective lead compound discovery by correlating phylogenetic, spatial, and bioactivity data. Proc. Natl. Acad. Sci. USA 2020, 117, 12444–12451. [Google Scholar] [CrossRef] [PubMed]
- Graf, B.A.; Milbury, P.E.; Blumberg, J.B. Flavonols, flavones, flavanones, and human health: Epidemiological evidence. J. Med. Food 2005, 8, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Arts, I.C.W.; Hollman, P.C.H. Polyphenols and disease risk in epidemiologic studies. Am. J. Clin. Nutr. 2005, 81. [Google Scholar] [CrossRef] [PubMed]
- Artasensi, A.; Pedretti, A.; Vistoli, G.; Fumagalli, L. Type 2 Diabetes Mellitus: A Review of Multi-Target Drugs. Molecules 2020, 25, 1987. [Google Scholar] [CrossRef] [PubMed]
- Baron, G.; Altomare, A.; Mol, M.; Garcia, J.L.; Correa, C.; Raucci, A.; Mancinelli, L.; Mazzotta, S.; Fumagalli, L.; Trunfio, G.; et al. Analytical profile and antioxidant and anti-inflammatory activities of the enriched polyphenol fractions isolated from bergamot fruit and leave. Antioxidants 2021, 10, 141. [Google Scholar] [CrossRef] [PubMed]
- Baron, G.; Altomare, A.; Regazzoni, L.; Fumagalli, L.; Artasensi, A.; Borghi, E.; Ottaviano, E.; Del Bo, C.; Riso, P.; Allegrini, P.; et al. Profiling Vaccinium macrocarpon components and metabolites in human urine and the urine ex-vivo effect on Candida albicans adhesion and biofilm-formation. Biochem. Pharmacol. 2020, 173, 113726. [Google Scholar] [CrossRef] [PubMed]
- Bross, M.; Herrmann, R.; Mocek, U. Monika Bross, Rupert Herrmann, Ursula Mocek. J. Antibiot. 1990, 43, 1413–1420. [Google Scholar]
- Teixeira, R.R.; Barbosa, L.C.A.; Maltha, C.R.A.; Rocha, M.E.; Bezerra, D.P.; Costa-Lotufo, L.V.; Pessoa, C.; Moraes, M.O. Synthesis and cytotoxic activity of some 3-benzyl-5-arylidenefuran-2(5H)-ones. Molecules 2007, 12, 1101–1116. [Google Scholar] [CrossRef] [PubMed]
- Sikorska, J.; Parker-Nance, S.; Davies-Coleman, M.T.; Vining, O.B.; Sikora, A.E.; McPhail, K.L. Antimicrobial rubrolides from a South African species of synoicum tunicate. J. Nat. Prod. 2012, 75, 1824–1827. [Google Scholar] [CrossRef] [PubMed]
- Negishi, E.-I.; Kotora, M. Tetrahedron Report Number 420 Regio-and Stereoselective Synthesis of y-Aikylidenebutenolides and Related Compounds; Elsevier: Amsterdam, The Netherlands, 1997; Volume 53. [Google Scholar]
- Sundar, N.; Kundu, M.K.; Reddy, P.V.; Mahendra, G.; Bhat, S.V. Zeolite mediated stereoselective synthesis of γ-alkylidenebutenolides. Synth. Commun. 2002, 32, 1881–1886. [Google Scholar] [CrossRef]
- Brückner, R. The β-elimination route to stereodefined γ-alkylidenebutenolides. Chem. Commun. 2001, 141–152. [Google Scholar] [CrossRef]
- Vaz, B.; Alvarez, R.; Brückner, R.; De Lera, A.R. The stille reaction in the synthesis of carotenoid butenolides: Synthesis of S′-epi-pridinin. Org. Lett. 2005, 7, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Boukouvalas, J.; Beltrán, P.P.; Lachance, N.; Côté, S.; Maltais, F.; Pouliot, M. A new, highly stereoselective synthesis of β-unsubstituted (Z)-γ-alkylidene-butenolides using bromine as a removable stereocontrol element. Synlett 2007, 2007, 219–222. [Google Scholar] [CrossRef]
- Casiraghi, G.; Battistini, L.; Curti, C.; Rassu, G.; Zanardi, F. The vinylogous aldol and related addition reactions: Ten years of progress. Chem. Rev. 2011, 111, 3076–3154. [Google Scholar] [CrossRef] [PubMed]
- Jefford, C.W.; Jaggi, D.; Boukouvalas, J. Diastereoselectivity in the directed aldol condensation of 2-trimethylsiloxyfuran with aldehydes. A stereodivergent route to threo and erythro δ-hydroxy-γ-lactones. Tetrahedron Lett. 1987, 28, 4037–4040. [Google Scholar] [CrossRef]
- Chang, X.; Peng, W.; Yao, Y.F.; Koek, J. Concise synthesis of ring-fission metabolites of epicatechin: 5-(3,4-Dihydroxybenzyl)dihydrofuran-2(3H)-one M6. Synth. Commun. 2010, 40, 3346–3352. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





