
4,5,6-Trichloropyrimidine-2-carboxamide



{kind=link}
{kind=link}
{kind=link}
Abstract
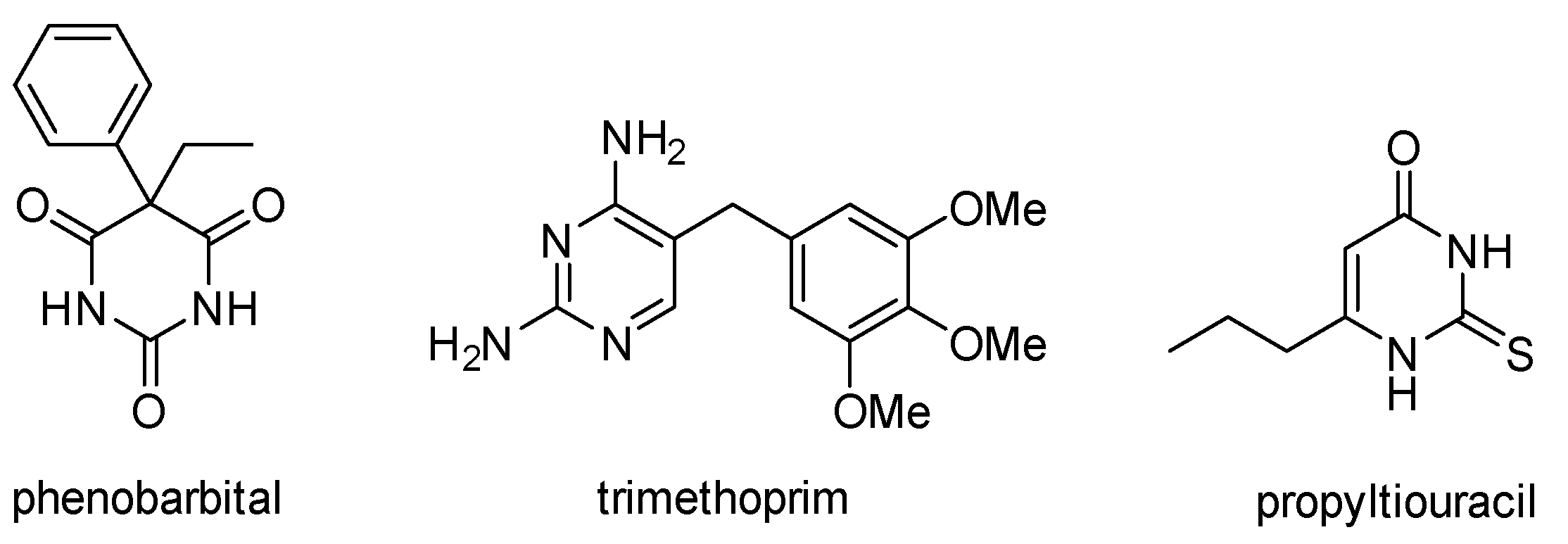
1. Introduction
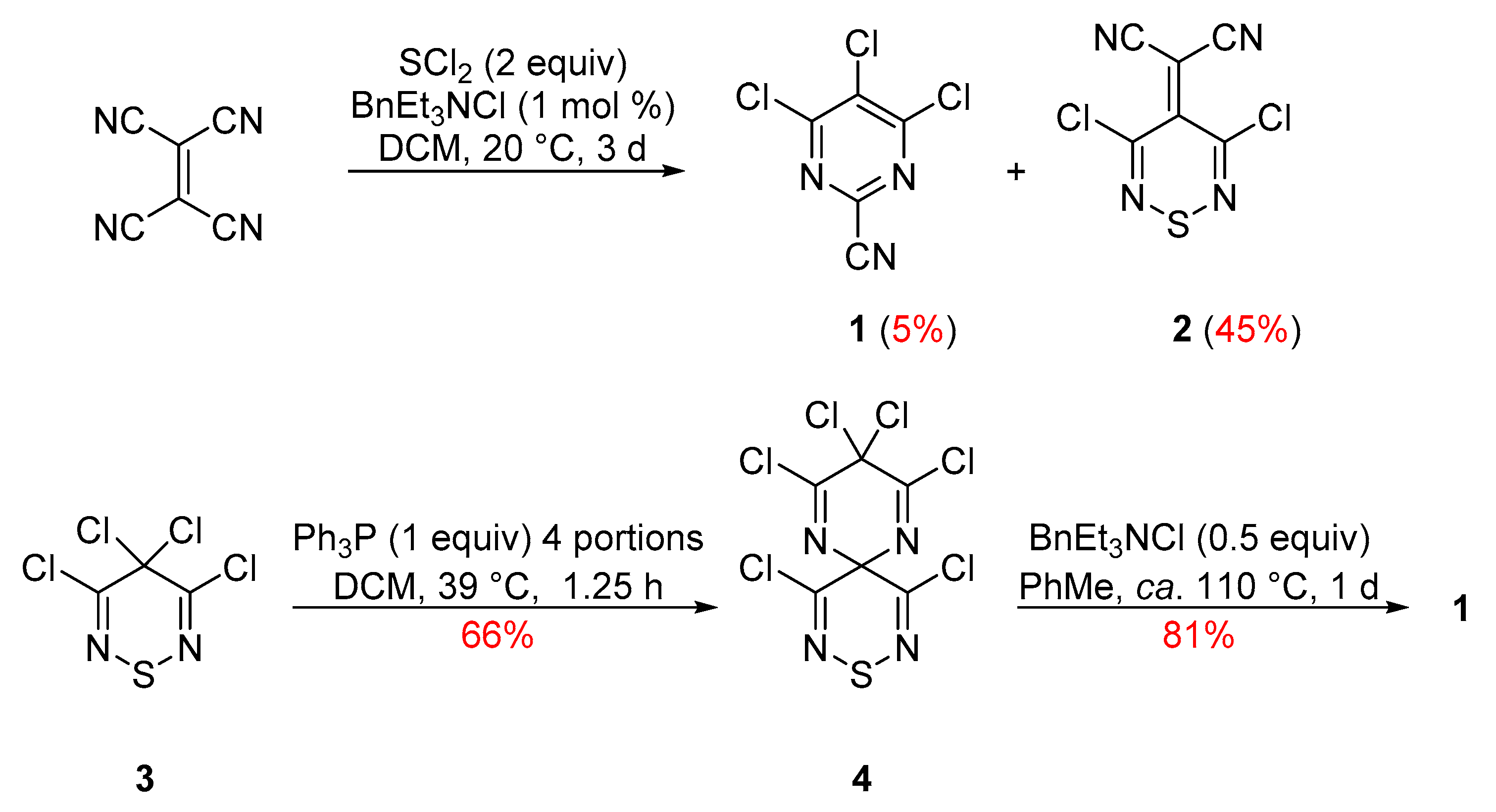
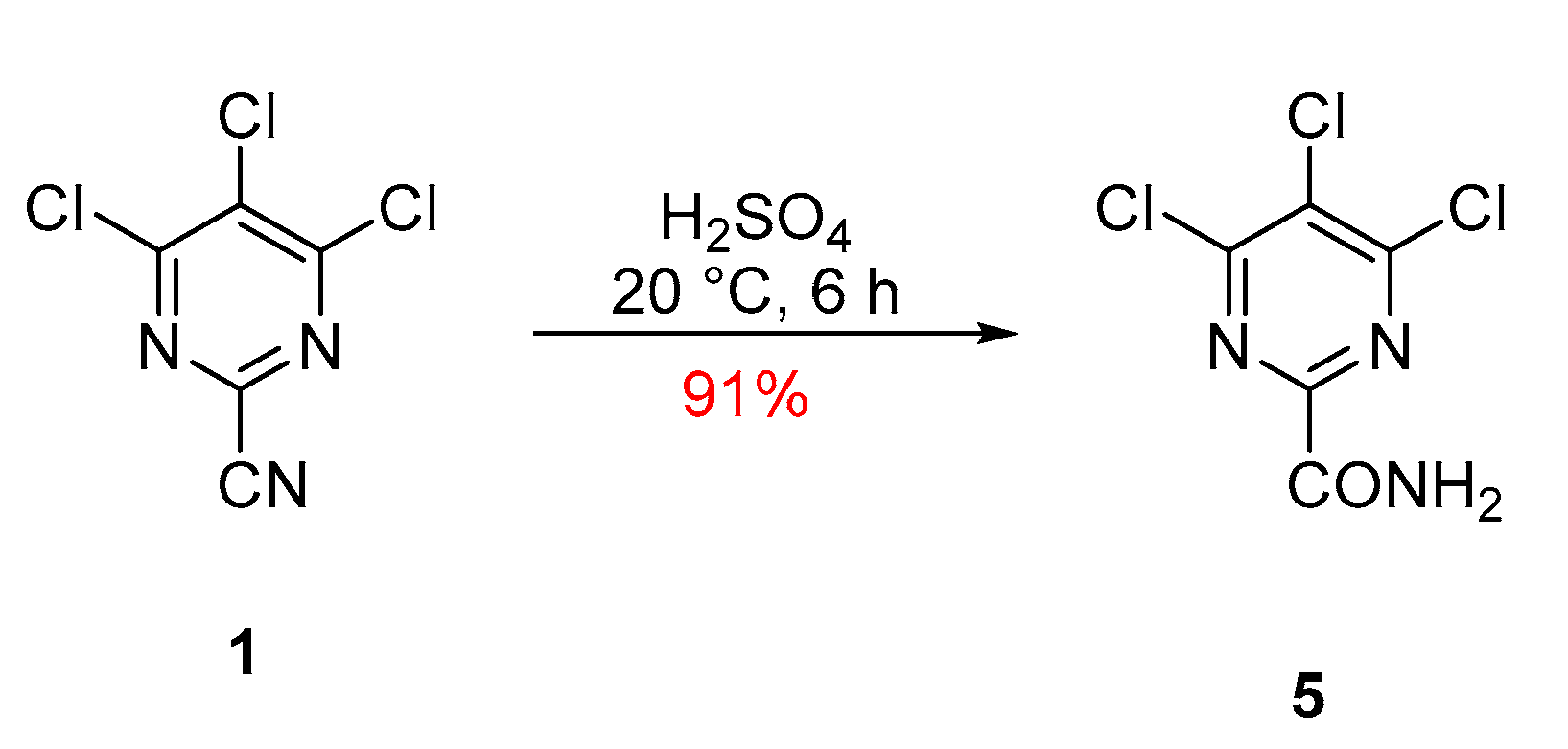
2. Results and Discussion
3. Materials and Methods
4,5,6-Trichloropyrimidine-2-carboxamide (5)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Soukup, G.A. Nucleic Acids: General Properties. In Encyclopedia of Life Sciences; Maccarrone, M., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2003. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Ukrainets, I.V.; Tugaibei, I.A.; Bereznykova, N.L.; Karvechenko, V.N.; Turov, A.V. 4-Hydroxy-2-quinolones 144. Alkyl-, arylalkyl-, and arylamides of 2-hydroxy-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid and their diuretic properties. Chem. Heterocycl. Com. 2008, 44, 565–575. [Google Scholar] [CrossRef]
- Amr, A.E.; Nermien, M.S.; Abdulla, M.M. Synthesis, reactions, and anti-inflammatory activity of heterocyclic systems fused to a thiophene moiety using citrazinic acid as synthon. Monatsh. Chem. 2007, 138, 699–707. [Google Scholar] [CrossRef]
- Gorlitzer, K.; Herbig, S.; Walter, R.D. Indeno[1,2-d]pyrimidin-4-yl-amines. Pharmazie 1997, 52, 670–672. [Google Scholar]
- Wagner, E.; Al-Kadasi, K.; Zimecki, M.; Sawka-Dobrowolska, W. Synthesis and pharmacological screening of derivatives of isoxazolo[4,5-d]pyrimidine. Eur. J. Med. Chem. 2008, 43, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.J. Pyrimidines and their Benzo Derivatives. In Comprehensive Heterocyclic Chemistry; Katritzky, A.R., Rees, C.W., Eds.; Pergamon Press: Oxford, UK, 1984; Volume 3, pp. 57–155. [Google Scholar]
- Koutentis, P.A.; Rees, C.W. Reaction of tetracyanoethylene with SCl2; new molecular rearrangements. J. Chem. Soc. Perkin Trans. 1 2000, 1089–1094. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Manoli, M.; Koutentis, P.A. Two-step conversion of 3,4,4,5-tetrachloro-4H-1,2,6-thiadiazine into 4,5,6-trichloropyrimidine-2-carbonitrile. Tetrahedron Lett. 2017, 58, 2618–2621. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Kourtellaris, A.; Koutentis, P.A. Synthesis of 4,5,6-trichloropyrimidine-2-carbonitrile from 4,6-dichloro-2-(methylthio)pyrimidine. ARKIVOC 2020, vii, 27–35. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Koutentis, P.A. Synthesis of 2-Cyanopyrimidines. Molbank 2019, 2019, M1086. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Koutentis, P.A. Reactions of Polychlorinated Pyrimidines with DABCO. Molbank 2019, 2019, M1084. [Google Scholar] [CrossRef]
- Christoforou, I.C.; Kalogirou, A.S.; Koutentis, P.A. The preparation of dicyano-1,3,4-thiadiazole and tricyanothiazole via 1,2,3-dithiazole chemistry. Tetrahedron 2009, 65, 9967–9972. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalogirou, A.S.; Koutentis, P.A. 4,5,6-Trichloropyrimidine-2-carboxamide. Molbank 2021, 2021, M1190. https://doi.org/10.3390/M1190
Kalogirou AS, Koutentis PA. 4,5,6-Trichloropyrimidine-2-carboxamide. Molbank. 2021; 2021(1):M1190. https://doi.org/10.3390/M1190
Chicago/Turabian StyleKalogirou, Andreas S., and Panayiotis A. Koutentis. 2021. "4,5,6-Trichloropyrimidine-2-carboxamide" Molbank 2021, no. 1: M1190. https://doi.org/10.3390/M1190
APA StyleKalogirou, A. S., & Koutentis, P. A. (2021). 4,5,6-Trichloropyrimidine-2-carboxamide. Molbank, 2021(1), M1190. https://doi.org/10.3390/M1190