Formation of an Isomeric Mixture of Dienynes Instead of a Diallene


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
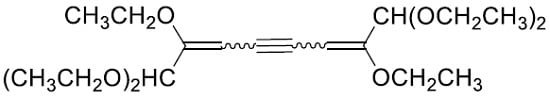
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Reduction of 1,1,2,2,7,7,8,8-Octaethoxyocta-3,5-diyne (1)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sydnes, L.K. Formation of Acetylenes by Ring Opening of 1,1,2-Trihalocyclopropanes. Eur. J. Org. Chem. 2000, 3511–3518. [Google Scholar] [CrossRef]
- Kvernenes, O.H.; Sydnes, L.K. Synthesis of 2-Chloroacrolein Diethyl Acetal (2-Chloroprop-2-enal, Diethyl Acetal. Org. Synth. 2005, 83, 184–192. [Google Scholar]
- Sydnes, L.K.; Holmelid, B.; Kvernenes, O.H.; Sandberg, M.; Hodne, M.; Bakstad, E. Synthesis and some chemical properties of 3,3,4,4-tetraethoxybut-1-yne. Tetrahedron 2007, 63, 4144–4148. [Google Scholar] [CrossRef]
- Sydnes, L.K.; Valdersnes, S. Recent advances in the synthesis of carbohydrate analogues. Pure Appl. Chem. 2007, 79, 2137–2142. [Google Scholar] [CrossRef]
- Sydnes, L.K.; Holmelid, B.; Myagmasuren, S.; Hanstein, M. New regiospecific synthesis of tri- and tetrasubstituted furans. J. Org. Chem. 2009, 74, 3430–3443. [Google Scholar] [CrossRef]
- Sydnes, L.K.; Isanov, R.; Sengee, M.; Livi, F. Regioselective Synthesis of Tetra-Substituted Furans. Synth. Commun. 2013, 43, 2898–2906. [Google Scholar] [CrossRef]
- Erdenebileg, U.; Høstmark, I.; Polden, K.; Sydnes, L.K. Synthesis and reactivity of 4-amino-sustituted furfurals. J. Org. Chem. 2014, 79, 1213–1221. [Google Scholar] [CrossRef]
- Farooq, T.; Haug, B.E.; Sydnes, L.K.; Törnroos, K.W. 1,3-Dipolar cycloaddition of benzyl azide to two highly functionalized alkynes. Monatshefte Chem. 2012, 143, 505–512. [Google Scholar] [CrossRef][Green Version]
- Sydnes, L.K.; Kvernenes, O.H.; Valdersnes, S. From 3,3,4,4-tetraethoxybutyne to carbohydrate mimics. Pure Appl. Chem. 2005, 77, 119–130. [Google Scholar] [CrossRef]
- Valdersnes, S.; Sydnes, L.K. Preparation of 2-ethoxy-3-hydroxy-4-(perfluoroalkyl)tetrahydropyran derivatives from substituted 4-ethoxybut-3-en-1-ols. Eur. J. Org. Chem. 2009, 5816–5831. [Google Scholar] [CrossRef]
- Sengee, M.; Sydnes, L.K. Specific conjugate addition to α,β-acetylenic ketones. Synthesis 2011, 23, 3899–3907. [Google Scholar] [CrossRef]
- Valdersnes, S.; Apeland, I.; Flemmen, G.; Sydnes, L.K. Toward the synthesis of modified carbohydrates by conjugate addition of propane-1,3-dithiol to α,β-unsaturated ketones. Helv. Chim. Acta 2012, 95, 2099–2122. [Google Scholar] [CrossRef]
- Leiren, M.K.; Valdersnes, S.; Sydnes, L.K. Selective transformations of a diprotected 2-oxo-butanedial. Helv. Chim. Acta 2013, 96, 1841–1850. [Google Scholar] [CrossRef]
- Nes, I.; Sydnes, L.K. Formation of N-heterocycles from 1,1-diethoxy-5-hydroxyalk-3-yn-2-ones. Synthesis 2014, 47, 89–94. [Google Scholar]
- Isanov, R.; Holmelid, B.; Törnroos, K.W.; Sydnes, L.K. Synthesis of (E)-1,1-diethoxy-3-(3-hydroxy-3-arylfuro [2,3-b]quinoxalin-2(3H)-ylidene)propan-2-ones via acid-catalyzed stereoselective 5-Exo-Dig cyclization. J. Heterocycl. Chem. 2015, 52, 711–718. [Google Scholar] [CrossRef]
- Holmelid, B.; Sydnes, L.K. Synthesis of 1,1,2,2,7,7,8,8-Octaethoxyocta-3,5-diyne. Molbank 2015, 2015, M840. [Google Scholar] [CrossRef]
- Nantz, M.H.; Bender, D.M.; Janaki, S. A convenient terminal allene synthesis from propargylic acetates. Synthesis 1993, 6, 577–578. [Google Scholar] [CrossRef]
- Haces, A.; Van Kruchten, E.M.G.A.; Okamura, W.H. On the stereochemistry of organocopper mediated conversion of propargylic esters to allenes. Israel J. Chem. 1985, 26, 140–146. [Google Scholar] [CrossRef]
- Elsevier, C.J.; Stehouwer, P.M.; Westmijze, H.; Vermeer, P. Anti-stereoselectivity in the palladium(0)-catalyzed conversion of propargylic esters into allenes by phenylzinc chloride. J. Org. Chem. 1983, 48, 1103–1105. [Google Scholar] [CrossRef]
- Mae, M.; Hong, J.A.; Xu, B.; Hammond, G.B. Highly Regioselective Synthesis of gem-Difluoroallenes through Magnesium Organocuprate SN2′ Substitution. Org. Lett. 2006, 8, 479–482. [Google Scholar] [CrossRef]
- Soler-Yanes, R.; Arribas-Alvarez, I.; Guisan-Ceinos, M.; Bunuel, E.; Cardenas, D.J. NiI Catalyzes the Regioselective Cross-Coupling of Alkylzinc Halides and Propargyl Bromide to Allenes. Chem. Eur. J. 2017, 23, 1584–1590. [Google Scholar] [CrossRef] [PubMed]
- Kalli, M.; Landor, P.D.; Landor, S.R. Allenes. XVII. Reaction of dialkyl(lithio)copper reagents with 1-bromoallenes, 1-iodoallenes, and 3-chloro-1-alkynes. J. Chem. Soc. Perkin Trans. 1 1973, 1347–1349. [Google Scholar] [CrossRef]
- Jacobs, T.L.; Wilcox, R.D. Dehalogenation of Propargyl and Allenyl Halides. II. J. Am. Chem. Soc. 1964, 86, 2240–2247. [Google Scholar] [CrossRef]
- Cowie, J.S.; Landor, P.D.; Landor, S.R. Allenes. XIV. Preparation of α-allenic alcohols from the mono-O-(tetrahydropyran-2-yl) derivatives of 1,4-butynediols. J. Chem. Soc. Perkin Trans. 1 1973, 720–724. [Google Scholar] [CrossRef]
- Alexakis, A.; Marek, I.; Mageney, P.; Normant, J.F. Mechanistic Aspects on the Formation of Chiral Allenes from Propargylic Ethers and Organocopper Reagents. J. Am. Chem. Soc. 1990, 112, 8042–8047. [Google Scholar] [CrossRef]
- Alexakis, A. Stereochemical Aspects on the Formation of Chiral Allenes from Propargylic Ethers and Epoxides. Pure Appl. Chem. 1992, 64, 387–392. [Google Scholar] [CrossRef]
- Boreux, A.; Lonca, G.H.; Riant, O.; Gagosz, F. Synthesis of Trifluoromethylallenes by Gold-Catalyzed Rearrangement Propargyl Benzyl Ethers. Org. Lett. 2016, 18, 5162–5165. [Google Scholar] [CrossRef]
- Bellamy, L.J. The Infrared Spectra of Complex Molecules, 2nd ed.; Chapman and Hall: London, UK, 1980; Volume 2. [Google Scholar]
- Becker, E.D. High Resolution NMR: Theory and Applications, 3rd ed.; Academic Press: San Diego, CA, USA, 2000; pp. 83–117. [Google Scholar]
- Skatteboel, L.; Solomon, S. Thermally induced reactions of some novel allenes. J. Am. Chem. Soc. 1965, 87, 4506–4513. [Google Scholar] [CrossRef]
- Mühlstädt:, M.; Graefe, J. Synthesen cycloaliphatischer Ketone aus Cyclodecatrien-(1,5,9). II. Herstellung von Cyclononen durch Hydratisierung cyclischer Allene. Chem. Ber. 1967, 100, 223–227. [Google Scholar] [CrossRef]
- Hoff, S.; Brandsma, L.; Arens, J.F. Conversions of allenyl ethers. Rec. Trav. Chim. Pay-Bas 1968, 87, 1179–1184. [Google Scholar] [CrossRef]
- Pasto, D.J.; Kong, W. Study of the substituted vinylallene-methylenecyclobutene electrocyclic equilibria. Comparison with the butadiene-cyclobutene and bisallene-bismethylenecyclobutene electrocyclic equilibria. J. Org. Chem. 1989, 54, 4028–4033. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrova, S.M.; Sydnes, L.K. Formation of an Isomeric Mixture of Dienynes Instead of a Diallene. Molbank 2020, 2020, M1133. https://doi.org/10.3390/M1133
Petrova SM, Sydnes LK. Formation of an Isomeric Mixture of Dienynes Instead of a Diallene. Molbank. 2020; 2020(2):M1133. https://doi.org/10.3390/M1133
Chicago/Turabian StylePetrova, Susanne M., and Leiv K. Sydnes. 2020. "Formation of an Isomeric Mixture of Dienynes Instead of a Diallene" Molbank 2020, no. 2: M1133. https://doi.org/10.3390/M1133
APA StylePetrova, S. M., & Sydnes, L. K. (2020). Formation of an Isomeric Mixture of Dienynes Instead of a Diallene. Molbank, 2020(2), M1133. https://doi.org/10.3390/M1133