Unexpected Seven-Membered Ring Formation for Muraymycin-Type Nucleoside-Peptide Antibiotics


Abstract
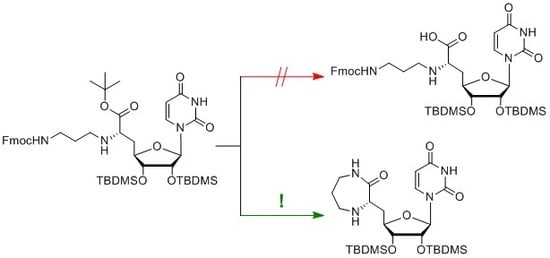
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Synthesis
4.2. Overexpression of MraY from S. aureus
4.3. Fluorescence-Based MraY Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Taubes, G. The bacteria fight back. Science 2008, 321, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A.; Shlaes, D. Fix the antibiotics pipeline. Nature 2011, 472, 32. [Google Scholar] [CrossRef] [PubMed]
- Von Nussbaum, F.; Brands, M.; Hinzen, B.; Weigand, S.; Häbich, D. Antibacterial Natural Products in Medicinal Chemistry - Exodus or Revival? Angew. Chem. Int. Ed. 2006, 45, 5072–5129. [Google Scholar] [CrossRef]
- Walsh, C. Where will new antibiotics come from? Nat. Rev. Microbiol. 2003, 1, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.; Goss, R.J.M.; Kimura, K.; Bugg, T.D.H. Antimicrobial nucleoside antibiotics targeting cell wall assembly: Recent advances in structure–function studies and nucleoside biosynthesis. Nat. Prod. Rep. 2010, 27, 279–304. [Google Scholar] [CrossRef]
- Wiegmann, D.; Koppermann, S.; Wirth, M.; Niro, G.; Leyerer, K.; Ducho, C. Muraymycin nucleoside-peptide antibiotics: Uridine-derived natural products as lead structures for the development of novel antibacterial agents. Beilstein J. Org. Chem. 2016, 12, 769–795. [Google Scholar] [CrossRef]
- Ichikawa, S.; Yamaguchi, M.; Matsuda, A. Antibacterial Nucleoside Natural Products Inhibiting Phospho-MurNAc-Pentapeptide Translocase; Chemistry and Structure–activity Relationship. Curr. Med. Chem. 2015, 22, 3951–3979. [Google Scholar] [CrossRef]
- Struve, W.G.; Neuhaus, F.C. Evidence for an Initial Acceptor of UDP-NAc-Muramyl-Pentapeptide in the Synthesis of Bacterial Mucopeptide. Biochem. Biophys. Res. Commun. 1965, 18, 6–12. [Google Scholar] [CrossRef]
- Anderson, J.S.; Matsuhashi, M.; Haskin, M.A.; Strominger, J.L. Lipid-Phosphoacetylmuramyl-pentapeptide and Lipid-Phosphodisaccharide-pentapeptide: Presumed Membrane Transport Intermediates in Cell Wall Synthesis. Proc. Natl. Acad. Sci. USA 1965, 53, 881–889. [Google Scholar] [CrossRef]
- Heydanek, M.G., Jr.; Struve, W.G.; Neuhaus, F.C. Initial state in peptidoglycan synthesis. III. Kinetics and uncoupling of phospho-N-acetylmuramyl-pentapeptide translocase. Biochemistry 1969, 8, 1214–1221. [Google Scholar] [CrossRef]
- Ikeda, M.; Wachi, M.; Jung, H.K.; Ishino, F.; Matsuhashi, M. The Escherichia coli mraY gene encoding UDP-N-acetylmuramoyl-pentapeptide: Undecaprenyl-phosphate phospho-N-acetylmuramoyl-pentapeptide transferase. J. Bacteriol. 1991, 173, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Boyle, D.S.; Donachie, W.D. mraY is an essential gene for cell growth in Escherichia coli. J. Bacteriol. 1998, 180, 6429–6432. [Google Scholar] [CrossRef] [PubMed]
- Bouhss, A.; Mengin-Lecreulx, D.; Le Beller, D.; Van Heijenoort, J. Topological analysis of the MraY protein catalysing the first membrane step of peptidoglycan synthesis. Mol. Microbiol. 1999, 34, 576–585. [Google Scholar] [CrossRef] [PubMed]
- McDonald, L.A.; Barbieri, L.R.; Carter, G.T.; Lenoy, E.; Lotvin, J.; Petersen, P.J.; Siegel, M.M.; Singh, G.; Williamson, R.T. Structures of the Muraymycins, Novel Peptidoglycan Biosynthesis Inhibitors. J. Am. Chem. Soc. 2002, 124, 10260–10261. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Wang, X.; Koppermann, S.; Thorson, J.S.; Ducho, C.; Van Lanen, S.G. Antibacterial Muraymycins from Mutant Strains of Streptomyces sp. NRRL 30471. J. Nat. Prod. 2018, 81, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Nakagawa, N.; Doi, N.; Hattori, S.; Naganawa, H.; Hamada, M. Caprazamycin B, a Novel Anti-tuberculosis Antibiotic, from Streptomyces sp. J. Antibiot. 2003, 56, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Takahashi, Y.; Shitara, T.; Nakamura, H.; Naganawa, H.; Miyake, T.; Akamatsu, Y. Caprazamycins, Novel Lipo-nucleoside Antibiotics, from Streptomyces sp. J. Antibiot. 2005, 58, 327–337. [Google Scholar] [CrossRef]
- Nakamura, H.; Tsukano, C.; Yasui, M.; Yokouchi, S.; Igarashi, M.; Takemoto, Y. Total Synthesis of (−)-Caprazamycin A. Angew. Chem. Int. Ed. 2015, 54, 3136–3139. [Google Scholar] [CrossRef]
- Abe, H.; Gopinath, P.; Ravi, G.; Wang, L.; Watanabe, T.; Shibasaki, M. Synthesis of caprazamycin B. Tetrahedron Lett. 2015, 56, 3782–3785. [Google Scholar] [CrossRef]
- Hirano, S.; Ichikawa, S.; Matsuda, A. Synthesis of Caprazamycin Analogues and Their Structure−Activity Relationship for Antibacterial Activity. J. Org. Chem. 2008, 73, 569–577. [Google Scholar] [CrossRef]
- Linder, R.; Ducho, C. Unified Synthesis of Densely Functionalized Amino Acid Building Blocks for the Preparation of Caprazamycin Nucleoside Antibiotics. Eur. J. Org. Chem. 2019, 2019, 1523–1534. [Google Scholar] [CrossRef]
- Sarabia, F.; Vivar-García, C.; García-Ruiz, C.; Martín-Ortiz, L.; Romero-Carrasco, A. Exploring the Chemistry of Epoxy Amides for the Synthesis of the 2′′-epi-Diazepanone Core of Liposidomycins and Caprazamycins. J. Org. Chem. 2012, 77, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.C.; Zhao, J.; Gillespie, R.A.; Kwon, D.-Y.; Guan, Z.; Hong, J.; Zhou, P.; Lee, S.-Y. Crystal Structure of MraY, an Essential Membrane Enzyme for Bacterial Cell Wall Synthesis. Science 2013, 341, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.C.; Mashalidis, E.H.; Tanino, T.; Kim, M.; Matsuda, A.; Hong, J.; Ichikawa, S.; Lee, S.-Y. Structural insights into inhibition of lipid I production in bacterial cell wall synthesis. Nature 2016, 533, 557–560. [Google Scholar] [CrossRef]
- Koppermann, S.; Ducho, C. Natural Products at Work: Structural Insights into Inhibition of the Bacterial Membrane Protein MraY. Angew. Chem. Int. Ed. 2016, 55, 11722–11724. [Google Scholar] [CrossRef]
- Yamashita, A.; Norton, E.; Petersen, P.J.; Rasmussen, B.A.; Singh, G.; Yang, Y.; Mansour, T.S.; Ho, D.M. Muraymycins, novel peptidoglycan biosynthesis inhibitors: Synthesis and SAR of their analogues. Bioorg. Med. Chem. Lett. 2003, 13, 3345–3350. [Google Scholar] [CrossRef]
- Tanino, T.; Ichikawa, S.; Al-Dabbagh, B.; Bouhss, A.; Oyama, H.; Matsuda, A. Synthesis and Biological Evaluation of Muraymycin Analogues Active against Anti-Drug-Resistant Bacteria. ACS Med. Chem. Lett. 2010, 1, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Tanino, T.; Al-Dabbagh, B.; Mengin-Lecreulx, D.; Bouhss, A.; Oyama, H.; Ichikawa, S.; Matsuda, A. Mechanistic Analysis of Muraymycin Analogues: A Guide to the Design of MraY Inhibitors. J. Med. Chem. 2011, 54, 8421–8439. [Google Scholar] [CrossRef]
- Takeoka, Y.; Tanino, T.; Sekiguchi, M.; Yonezawa, S.; Sakagami, M.; Takahashi, F.; Togame, H.; Tanaka, Y.; Takemoto, H.; Ichikawa, S.; et al. Expansion of Antibacterial Spectrum of Muraymycins toward Pseudomonas aeruginosa. ACS Med. Chem. Lett. 2014, 5, 556–560. [Google Scholar] [CrossRef]
- Spork, A.P.; Büschleb, M.; Ries, O.; Wiegmann, D.; Boettcher, S.; Mihalyi, A.; Bugg, T.D.; Ducho, C. Lead Structures for New Antibacterials: Stereocontrolled Synthesis of a Bioactive Muraymycin Analogue. Chem.- Eur. J. 2014, 20, 15292–15297. [Google Scholar] [CrossRef]
- Mitachi, K.; Aleiwi, B.A.; Schneider, C.M.; Siricilla, S.; Kurosu, M. Stereocontrolled Total Synthesis of Muraymycin D1 Having a Dual Mode of Action against Mycobacterium tuberculosis. J. Am. Chem. Soc. 2016, 138, 12975–12980. [Google Scholar] [CrossRef] [PubMed]
- Koppermann, S.; Cui, Z.; Fischer, P.D.; Wang, X.; Ludwig, J.; Thorson, J.S.; Van Lanen, S.G.; Ducho, C. Insights into the Target Interaction of Naturally Occurring Muraymycin Nucleoside Antibiotics. ChemMedChem 2018, 13, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Spork, A.P.; Koppermann, S.; Schier (née Wohnig), S.; Linder, R.; Ducho, C. Analogues of Muraymycin Nucleoside Antibiotics with Epimeric Uridine-Derived Core Structures. Molecules 2018, 23, 2868. [Google Scholar] [CrossRef] [PubMed]
- Wiegmann, D.; Koppermann, S.; Ducho, C. Aminoribosylated Analogues of Muraymycin Nucleoside Antibiotics. Molecules 2018, 23, 3085. [Google Scholar] [CrossRef] [PubMed]
- Heib, A.; Niro, G.; Weck, S.C.; Koppermann, S.; Ducho, C. Muraymycin Nucleoside Antibiotics: Structure–activity Relationship for Variations in the Nucleoside Unit. Molecules 2020, 25, 22. [Google Scholar] [CrossRef]
- Leyerer, K.; Koppermann, S.; Ducho, C. Solid Phase-Supported Synthesis of Muraymycin Analogues. Eur. J. Org. Chem. 2019, 45, 7420–7431. [Google Scholar] [CrossRef]
- Spork, A.P.; Ducho, C. Novel 5′-deoxy nucleosyl amino acid scaffolds for the synthesis of muraymycin analogues. Org. Biomol. Chem. 2010, 8, 2323–2326. [Google Scholar] [CrossRef]
- Spork, A.P.; Wiegmann, D.; Granitzka, M.; Stalke, D.; Ducho, C. Stereoselective Synthesis of Uridine-Derived Nucleosyl Amino Acids. J. Org. Chem. 2011, 76, 10083–10098. [Google Scholar] [CrossRef]
- Schmidtgall, B.; Höbartner, C.; Ducho, C. NAA-modified DNA oligonucleotides with zwitterionic backbones: Stereoselective synthesis of A–T phosphoramidite building blocks. Beilstein J. Org. Chem. 2015, 11, 50–60. [Google Scholar] [CrossRef]
- Wohnig, S.; Spork, A.P.; Koppermann, S.; Mieskes, G.; Gisch, N.; Jahn, R.; Ducho, C. Total Synthesis of Dansylated Park’s Nucleotide for High-Throughput MraY Assays. Chem. – Eur. J. 2016, 22, 17813–17819. [Google Scholar] [CrossRef]
- Kaysser, L.; Lutsch, L.; Siebenberg, S.; Wemakor, E.; Kammerer, B.; Gust, B. Identification and Manipulation of the Caprazamycin Gene Cluster Lead to New Simplified Liponucleoside Antibiotics and Give Insights into the Biosynthetic Pathway. J. Biol. Chem. 2009, 284, 14987–14996. [Google Scholar] [CrossRef] [PubMed]
- Wiker, F.; Hauck, N.; Grond, S.; Gust, B. Caprazamycins: Biosynthesis and structure activity relationship studies. Int. J. Med. Microbiol. 2019, 309, 319–324. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Reagents | Temperature | Reaction Period | Product |
|---|---|---|---|---|
| 1 | SiO2, toluene | 111 °C (reflux) | 5 days | 41% 6 |
| 2 | SiO2, toluene | 111 °C (reflux) | 1.5 days | 68% 6 |
| 3 | SiO2, toluene | 111 °C (reflux) | 5 h | 76% 6 |
| 4 | SiO2, toluene | 80 °C, then rt | 3 days | 48% 5 |
| 5 | 80% TFA/CH2Cl2 | rt | 2 h | partial TBDMS deprotection |
| 6 | 80% TFA/CH2Cl2 | rt | 1 h | 16% 5 (24% brsm 1) |
| 7 | 50% TFA/CH2Cl2 | rt | 2.5 h | full TBDMS deprotection |
| 8 | PLE, phosphate buffer (pH 7.5, 0.1 M), DMF | rt | 2 days | no reaction |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leyerer, K.; Koppermann, S.; Ducho, C. Unexpected Seven-Membered Ring Formation for Muraymycin-Type Nucleoside-Peptide Antibiotics. Molbank 2020, 2020, M1122. https://doi.org/10.3390/M1122
Leyerer K, Koppermann S, Ducho C. Unexpected Seven-Membered Ring Formation for Muraymycin-Type Nucleoside-Peptide Antibiotics. Molbank. 2020; 2020(2):M1122. https://doi.org/10.3390/M1122
Chicago/Turabian StyleLeyerer, Kristin, Stefan Koppermann, and Christian Ducho. 2020. "Unexpected Seven-Membered Ring Formation for Muraymycin-Type Nucleoside-Peptide Antibiotics" Molbank 2020, no. 2: M1122. https://doi.org/10.3390/M1122
APA StyleLeyerer, K., Koppermann, S., & Ducho, C. (2020). Unexpected Seven-Membered Ring Formation for Muraymycin-Type Nucleoside-Peptide Antibiotics. Molbank, 2020(2), M1122. https://doi.org/10.3390/M1122