(E)-(1-(4-Ethoxycarbonylphenyl)-5-(3,4-dimethoxyphenyl)-3-(3,4-dimethoxystyryl)-2-pyrazoline: Synthesis, Characterization, DNA-Interaction, and Evaluation of Activity Against Drug-Resistant Cell Lines

 ,
,  and
and
Abstract
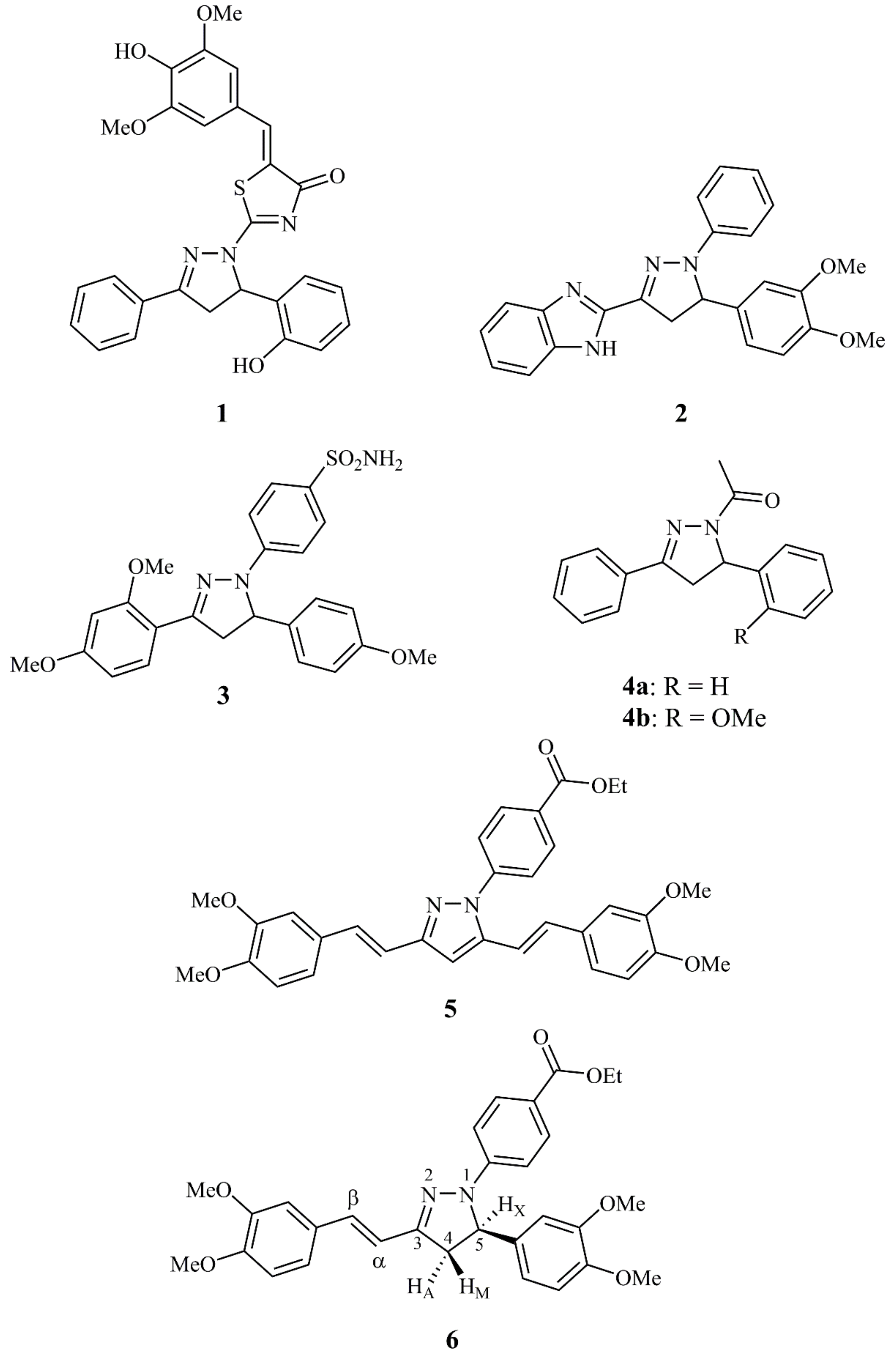
1. Introduction
2. Results and Discussion
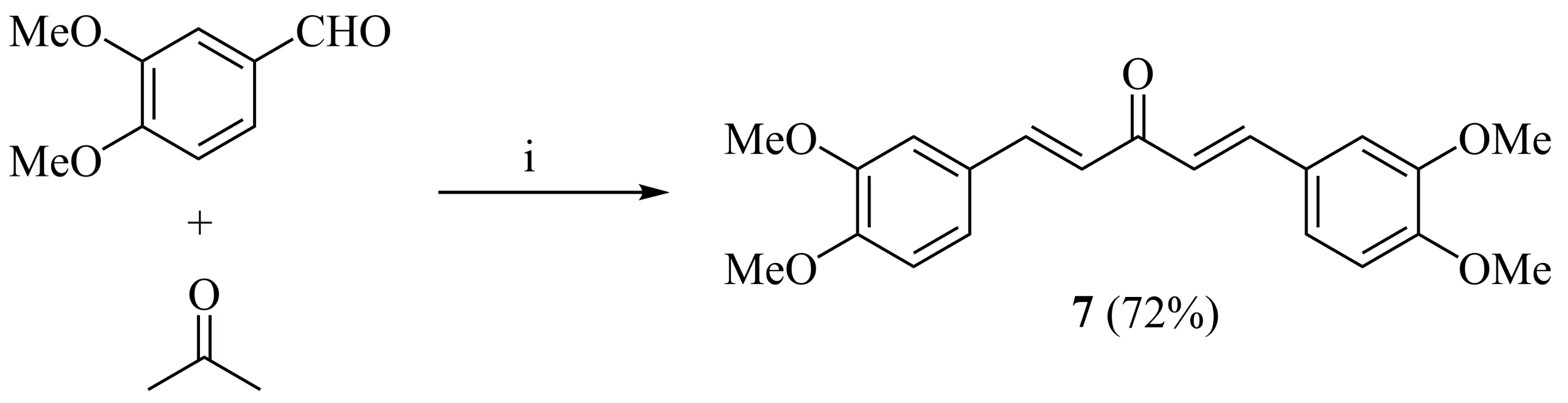
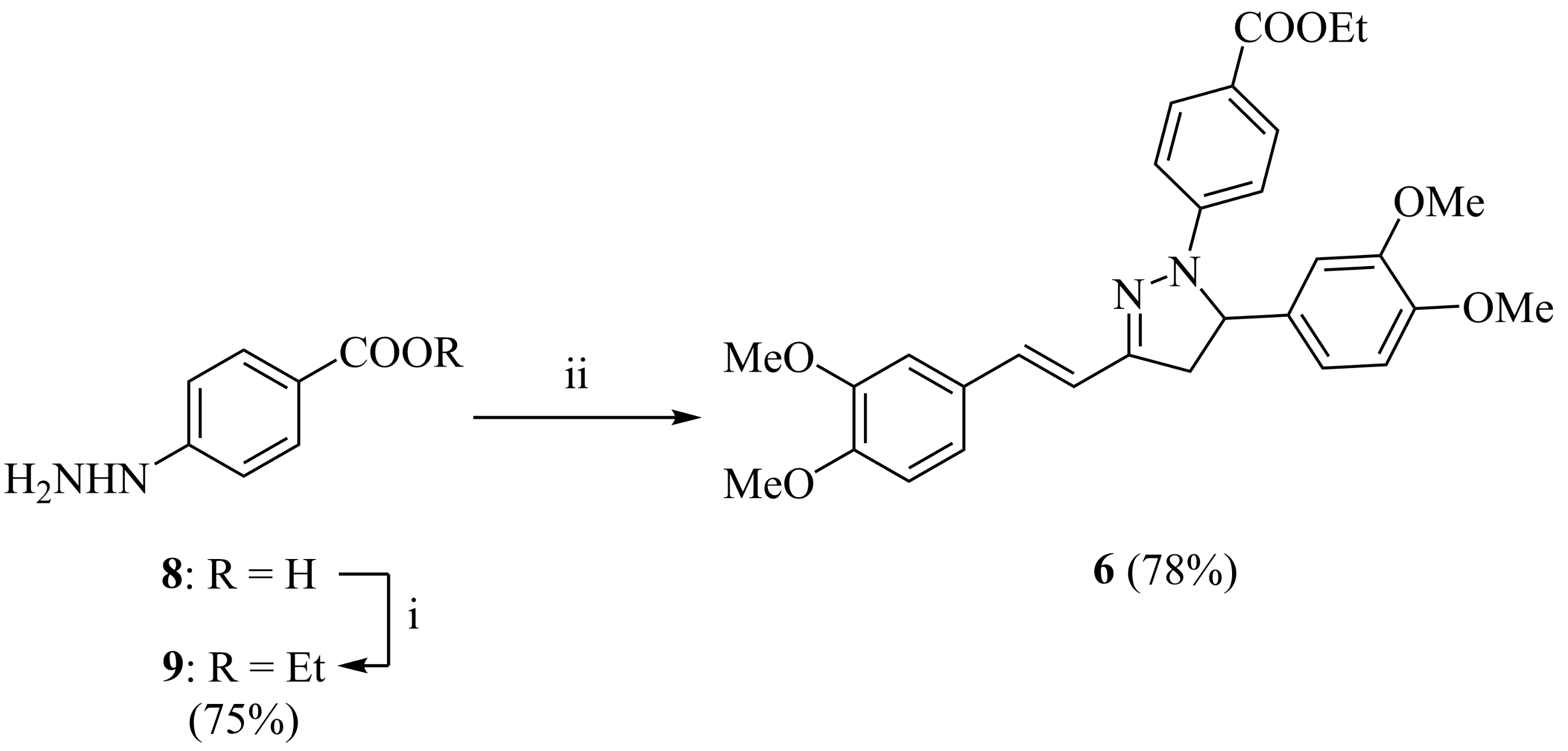
2.1. Synthesis and Characterization
2.2. Biological Evaluation
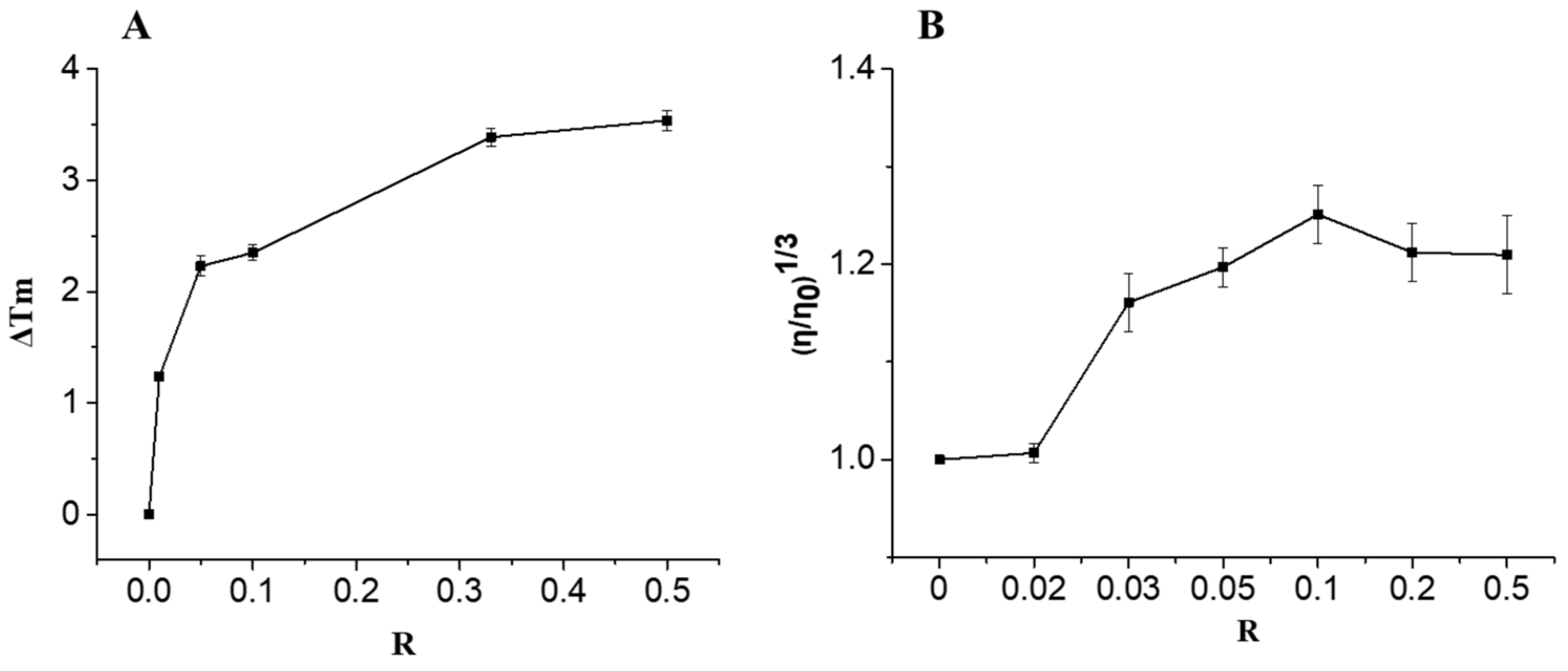
2.2.1. DNA Binding Studies
2.2.2. In Vitro Cytotoxicity Studies Against Doxorubicin-Resistant Breast Cancer Cells
3. Materials and Methods
3.1. General
3.2. (1E,4E)-1,5-Bis(3,4-dimethoxyphenyl)penta-1,4-dien-3-one (7)
3.3. Ethyl 4-hydrazinobenzoate (9)
3.4. (E)-1-(4-Ethoxycarbonylphenyl)-5-(3,4-dimethoxyphenyl)-3-(3,4-dimethoxystyryl)-2-pyrazoline (6)
3.5. Preparation of DNA Samples
3.6. Thermal Denaturation Studies
3.7. Viscosity Studies
3.8. Cell Culture
3.9. In Vitro Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Natural Products in Medicinal Chemistry; Hanessian, S., Ed.; Wiley-VCH:: Weinheim, Germany, 2014. [Google Scholar]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Varghese, B.; Al-Busafi, S.N.; Suliman, F.O.; Al-Kindy, M.Z. Unveiling a versatile heterocycle: Pyrazoline—A review. RSC Adv. 2017, 7, 46999–47016. [Google Scholar] [CrossRef]
- Shaaban, M.R.; Mayhoub, A.S.; Farag, A.M. Recent advances in the therapeutic applications of pyrazolines. Expert Opim. Ther. Patents 2012, 22, 253–291. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, J.; Khan, A.A.; Ali, Z.; Haider, R.; Yar, M.S. Structure-activity relationship (SAR) study and design strategies of nitrogen-containing heterocyclic moieties for their anticancer activities. Eur. J. Med. Chem. 2017, 125, 143–189. [Google Scholar] [CrossRef] [PubMed]
- Havrylyuk, D.; Zimenkovsky, B.; Vasylenko, O.; Zaprutko, L.; Gzella, A.; Lesyk, R. Synthesis of novel thiazolone-based compounds containing pyrazoline moiety and evaluation of their anticancer activity. Eur. J. Med. Chem. 2009, 44, 1396–1404. [Google Scholar] [CrossRef] [PubMed]
- Shaharyar, M.; Abdullah, M.M.; Bakht, M.A.; Majeed, J. Pyrazoline bearing benzimidazoles: Search for anticancer agent. Eur. J. Med. Chem. 2010, 45, 114–119. [Google Scholar] [CrossRef]
- Insuasty, B.; Montoya, A.; Becerra, D.; Quiroga, J.; Abonia, R.; Robledo, S.; Vélez, I.D.; Upegui, Y.; Nogueras, M.; Cobo, J. Synthesis of novel analogs of 2-pyrazoline obtained from [(7-chloroquinolin-4-yl)amino]chalcones and hydrazine as potential antitumor and antimalarial agents. Eur. J. Med. Chem. 2013, 67, 252–262. [Google Scholar] [CrossRef]
- Shin, S.Y.; Yoon, H.; Hwang, D.; Ahn, S.; Kim, D.-W.; Koh, D.; Lee, Y.H.; Lim, Y. Benzochalcones bearing pyrazoline moieties show anti-colorectal cancer activities and selective inhibitory effects on aurora kinases. Bioorg. Med. Chem. 2013, 21, 7018–7024. [Google Scholar] [CrossRef]
- Rathish, I.G.; Javed, K.; Ahmad, S.; Bano, S.; Alam, M.S.; Pillai, K.K.; Singh, S.; Bagchi, V. Synthesis and antiinflammatory activity of some new 1,3,5-trisubstituted pyrazolines bearing benzene sulfonamide. Bioorg. Med. Chem. Lett. 2009, 19, 255–258. [Google Scholar] [CrossRef]
- Bansal, E.; Srivastava, V.K.; Kumar, A. Synthesis and anti-inflammatory activity of 1-acetyl-5-substitute daryl-3-(b-aminonaphthyl)-2-pyrazolines and b-(substituted aminoethyl) amidonaphthalenes. Eur. J. Med. Chem. 2001, 36, 81–92. [Google Scholar] [CrossRef]
- Lokeshwari, D.M.; Achutha, D.K.; Srinivasan, B.; Shivalingegowda, N.; Krishnappagowda, L.N.; Kariyappa, A.K. Synthesis of novel 2-pyrazoline analogues with potent anti-inflammatory effect mediated by inhibition of phospholipase A2: Crystallographic, in silico docking and QSAR analysis. Bioorg. Med. Chem. Lett. 2017, 27, 3806–3811. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, M.S.; Holla, B.S.; Kumari, N.S. Synthesis and antimicrobial studies on novel chloro-fluorine containing hydroxy pyrazolines. Eur. J. Med. Chem. 2007, 42, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, Z.N.; Musthafa, T.N.M.; Ahmad, A.; Khan, A.U. Thermal solvent-free synthesis of novel pyrazolyl chalcones and pyrazolines as potential antimicrobial agents. Bioorg. Med. Chem. Lett. 2011, 21, 2860–2865. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.K.; Mishra, M.; Kashaw, V.; Kashaw, S.K. Synthesis of 1,3,5-trisubstituted pyrazolines as potential antimalarial and antimicrobial agents. Bioorg. Med. Chem. 2017, 25, 1949–1962. [Google Scholar] [CrossRef] [PubMed]
- Manna, F.; Chimenti, F.; Fioravanti, R.; Bolasco, A.; Secci, D.; Chimenti, P.; Ferlini, C.; Scambia, G. Synthesis of some pyrazole derivatives and preliminary investigation of their affinity binding to P-glycoprotein. Bioorg. Med. Chem. Lett. 2005, 15, 4632–4635. [Google Scholar] [CrossRef]
- Kolotova, E.S.; Shtil, A.A.; Novikov, F.N.; Chilov, G.G.; Stroganov, O.V.; Stroilov, V.S.; Zeifman, A.A.; Titov, I.Y.; Sagnou, M.; Alexiou, P. Novel Derivatives of 3,5-Divinyl-pyrazole for Medical Application. WO 2016/190770 A1, 1 December 2016. [Google Scholar]
- Gottesman, M.M.; Pastan, I.; Ambudkar, S.V. P-Glycoprotein and multidrug resistance. Curr. Opin. Gen. Dev. 1996, 6, 610–617. [Google Scholar] [CrossRef]
- Halevas, E.; Mavroidi, B.; Swanson, C.H.; Smith, G.C.; Moschona, A.; Hadjispyrou, S.; Salifoglou, A.; Pantazaki, A.A.; Pelecanou, M.; Litsardakis, G. Magnetic cationic liposomal nanocarriers for the efficient drug delivery of a curcumin-based vanadium complex with anticancer potential. J. Inorg. Biochem. 2019, 199, 110778. [Google Scholar] [CrossRef]
- Mavroidi, B.; Sagnou, M.; Stamatakis, K.; Paravatou-Petsotas, M.; Pelecanou, M.; Methenitis, G. Palladium(II) and platinum(II) complexes of derivatives of 2-(4′-aminophenyl)benzothiazole as potential anticancer agents. Inorg Chim. Acta 2016, 444, 63–75. [Google Scholar] [CrossRef]
- Suh, D.; Chaires, J.B. Criteria for the mode of binding of DNA binding agents. Bioorg. Med. Chem. 1995, 3, 723–728. [Google Scholar] [CrossRef]
- Zsila, F.; Bikádi, Z.; Simonyi, M. Circular dichroism spectroscopic studies reveal pH dependent binding of curcumin in the minor groove of natural and synthetic nucleic acids. Org. Biomol. Chem. 2004, 2, 2902–2910. [Google Scholar]
- Kunwar, A.; Simon, E.; Singh, U.; Chittela, R.; Sharma, D.; Sandur, S.K.; Priyadarsini, I.K. Interaction of a curcumin analogue dimethoxycurcumin with DNA. Chem. Biol. Drug Des. 2011, 77, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, B.K.; Ghosh, K.S.; Bera, R.; Dasgupta, S. Studies on the interaction of diacetylcurcumin with calf thymus-DNA. Chem. Phys. 2008, 351, 163–169. [Google Scholar] [CrossRef]
- Bera, R.; Sahoo, B.K.; Ghosh, K.S.; Dasgupta, S. Studies on the interaction of isoxazolcurcumin with calf thymus DNA. Int. J. Biol. Macromol. 2008, 42, 14–21. [Google Scholar]
- Arif, I.S.; Hooper, C.L.; Greco, F.; Williams, A.C.; Boateng, S.Y. Increasing doxorubicin activity against breast cancer cells using PPARγ-ligands and by exploiting circadian rhythms. Br. J. Pharmacol. 2013, 169, 1178–1188. [Google Scholar] [CrossRef]
- Li, Q.; Chen, J.; Luo, S.; Xu, J.; Huang, Q.; Liu, T. Synthesis and assessment of the antioxidant and antitumor properties of asymmetric curcumin analogues. Eur. J. Med. Chem. 2015, 93, 461–469. [Google Scholar] [CrossRef]
- Butcher, R.J.; Jasinski, J.P.; Yathirajan, H.S.; Bindya, S.; Narayana, B.; Sarojini, B.K. 1,5-Bis(3,4-dimethoxyphenyl)penta-1,4-dien-3-one. Acta Cryst. 2007, E63, o3115. [Google Scholar] [CrossRef]
- Du, Z.-Y.; Bao, Y.-D.; Liu, Z.; Qiao, W.; Ma, L.; Huang, Z.-S.; Gu, L.-Q.; Chan, A.S.C. Curcumin analogs as potent aldose reductase inhibitors. Arch. Pharm. Chem. Life Sci. 2006, 339, 123–128. [Google Scholar] [CrossRef]
- Bourrie, B.; Casellas, P.; Ciapetti, P.; Derocq, J.-M.; Jegham, S.; Muneaux, Y.; Wermuth, C.-G. Pyridoindolone Derivatives Substituted in the 3-Position by a Phenyl, Their Preparation and Their Application in Therapeutics. US 7,390,818 B2, 24 June 2008. [Google Scholar]
- Cohen, G. Eisenber, Viscosity and sedimentation study of sonicated DNA–proflavine complexes. Biopolymers 1969, 8, 45. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) | |
|---|---|---|
| MCF-7 | MCF-7-DOXR | |
| 6 | 53.09 ± 3.58 | 85.11 ± 4.68 |
| DOX | 5.22 ± 1.33 | 70.90 ± 8.75 |
| DOX (pre-incubated with 6) | 7.71 ± 1.59 | 72.55 ± 4.99 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matiadis, D.; Mavroidi, B.; Panagiotopoulou, A.; Methenitis, C.; Pelecanou, M.; Sagnou, M. (E)-(1-(4-Ethoxycarbonylphenyl)-5-(3,4-dimethoxyphenyl)-3-(3,4-dimethoxystyryl)-2-pyrazoline: Synthesis, Characterization, DNA-Interaction, and Evaluation of Activity Against Drug-Resistant Cell Lines. Molbank 2020, 2020, M1114. https://doi.org/10.3390/M1114
Matiadis D, Mavroidi B, Panagiotopoulou A, Methenitis C, Pelecanou M, Sagnou M. (E)-(1-(4-Ethoxycarbonylphenyl)-5-(3,4-dimethoxyphenyl)-3-(3,4-dimethoxystyryl)-2-pyrazoline: Synthesis, Characterization, DNA-Interaction, and Evaluation of Activity Against Drug-Resistant Cell Lines. Molbank. 2020; 2020(1):M1114. https://doi.org/10.3390/M1114
Chicago/Turabian StyleMatiadis, Dimitris, Barbara Mavroidi, Angeliki Panagiotopoulou, Constantinos Methenitis, Maria Pelecanou, and Marina Sagnou. 2020. "(E)-(1-(4-Ethoxycarbonylphenyl)-5-(3,4-dimethoxyphenyl)-3-(3,4-dimethoxystyryl)-2-pyrazoline: Synthesis, Characterization, DNA-Interaction, and Evaluation of Activity Against Drug-Resistant Cell Lines" Molbank 2020, no. 1: M1114. https://doi.org/10.3390/M1114
APA StyleMatiadis, D., Mavroidi, B., Panagiotopoulou, A., Methenitis, C., Pelecanou, M., & Sagnou, M. (2020). (E)-(1-(4-Ethoxycarbonylphenyl)-5-(3,4-dimethoxyphenyl)-3-(3,4-dimethoxystyryl)-2-pyrazoline: Synthesis, Characterization, DNA-Interaction, and Evaluation of Activity Against Drug-Resistant Cell Lines. Molbank, 2020(1), M1114. https://doi.org/10.3390/M1114