3. Materials and Methods
The reaction mixture was monitored by TLC using commercial glass-backed thin-layer chromatography (TLC) plates (Merck Kieselgel 60 F
254). The plates were observed under UV light at 254 and 365 nm. Dichloromethane (DCM) and acetonitrile (MeCN) were distilled over CaH
2 before use. The 1 M and 3 M solutions of sodium methoxide (MeONa) in methanol (MeOH) were freshly prepared by the reaction of sodium metal with MeOH. The melting point was determined using a PolyTherm-A, Wagner & Munz, Kofler Hotstage Microscope apparatus (Wagner & Munz, Munich, Germany). The solvent used for recrystallization is indicated after the melting point. The UV-vis spectrum was obtained using a Perkin-Elmer Lambda-25 UV-vis spectrophotometer (Perkin-Elmer, Waltham, MA, USA), and inflections are identified by the abbreviation “inf”. The IR spectrum was recorded on a Shimadzu FTIR-NIR Prestige-21 spectrometer (Shimadzu, Kyoto, Japan) with Pike Miracle Ge ATR accessory (Pike Miracle, Madison, WI, USA) and strong, medium and weak peaks are represented by s, m and w, respectively.
1H and
13C NMR spectra were recorded on a Bruker Avance 500 spectrometer [at 500 and 125 MHz, respectively, (Bruker, Billerica, MA, USA)]. Deuterated solvents were used for homonuclear lock and the signals are referenced to the deuterated solvent peaks. Attached proton test (APT) NMR studies were used for the assignment of the
13C peaks as
CH
3,
CH
2,
CH, and
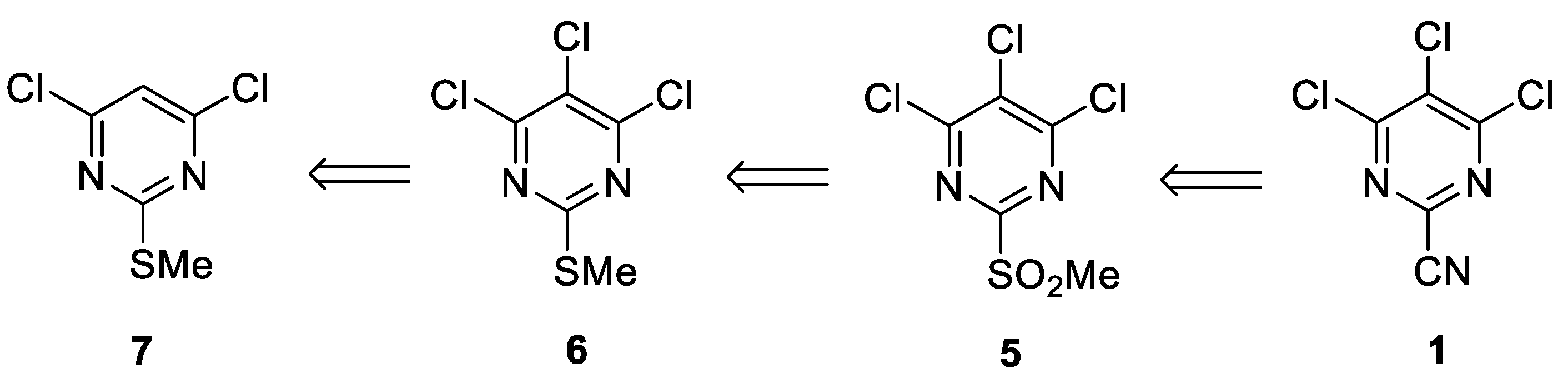
Cq (quaternary). The MALDI-TOF mass spectrum (+ve mode) was recorded on a Bruker Autoflex III Smartbeam instrument (Bruker). The elemental analysis was run by the London Metropolitan University Elemental Analysis Service. 4,6-dichloro-2-(methylthio)pyrimidine (
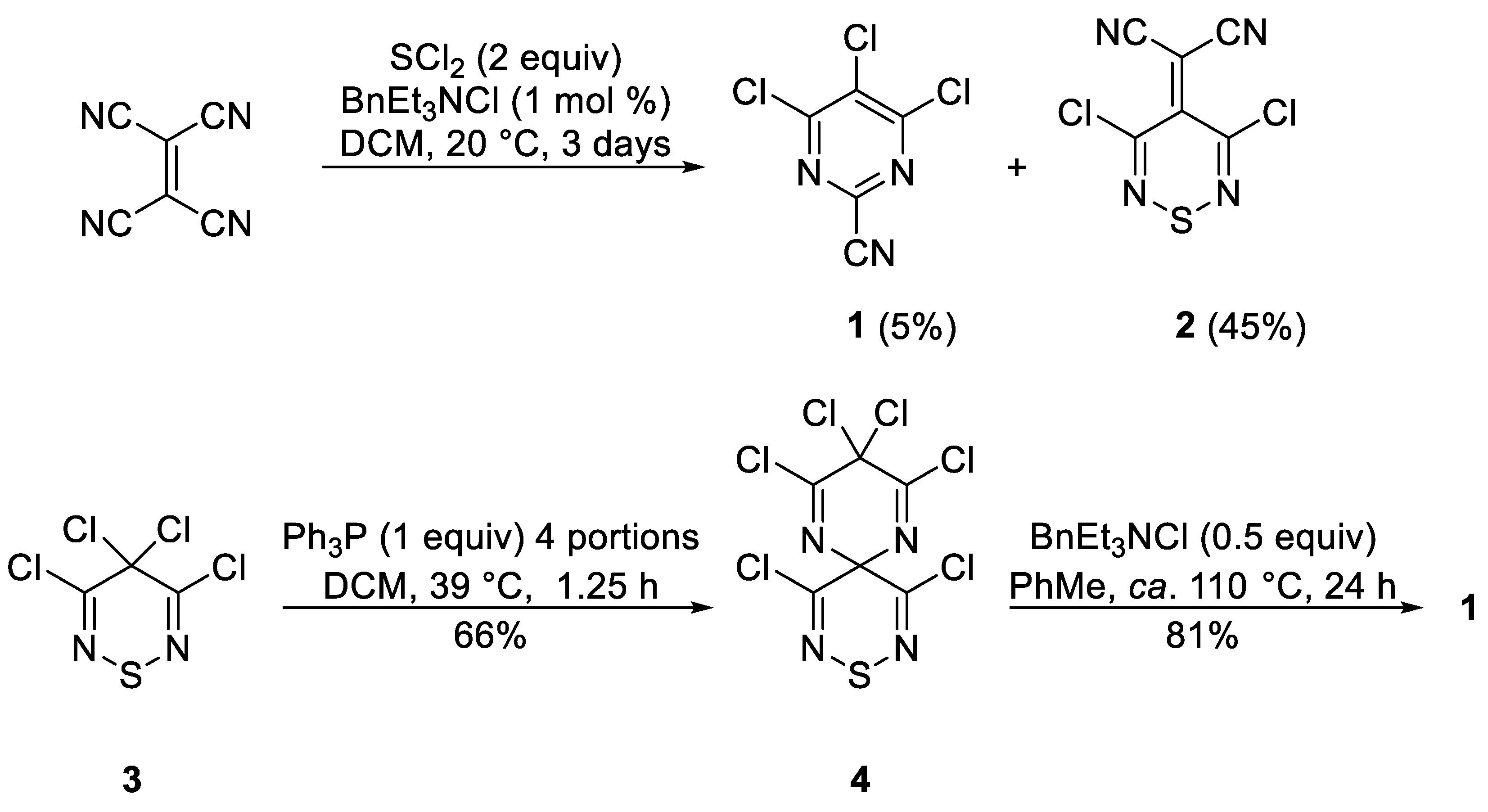
7) was prepared according to the literature procedure [
8].
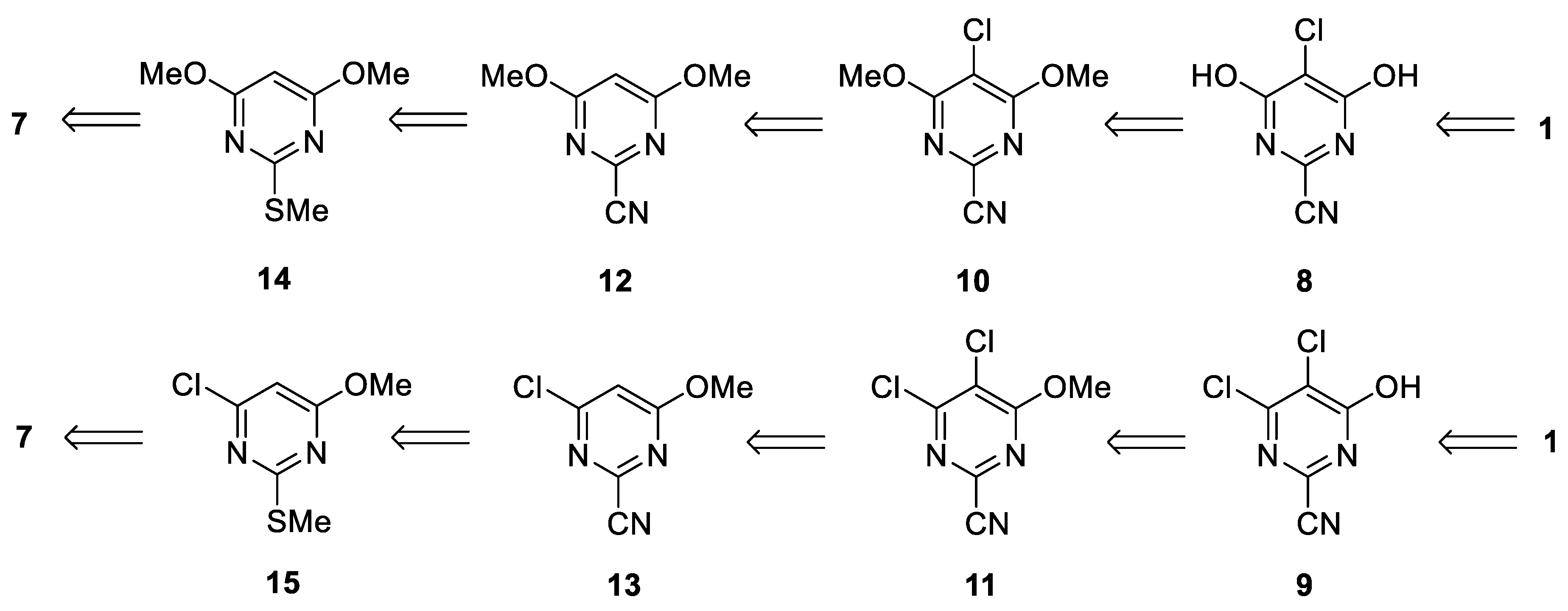
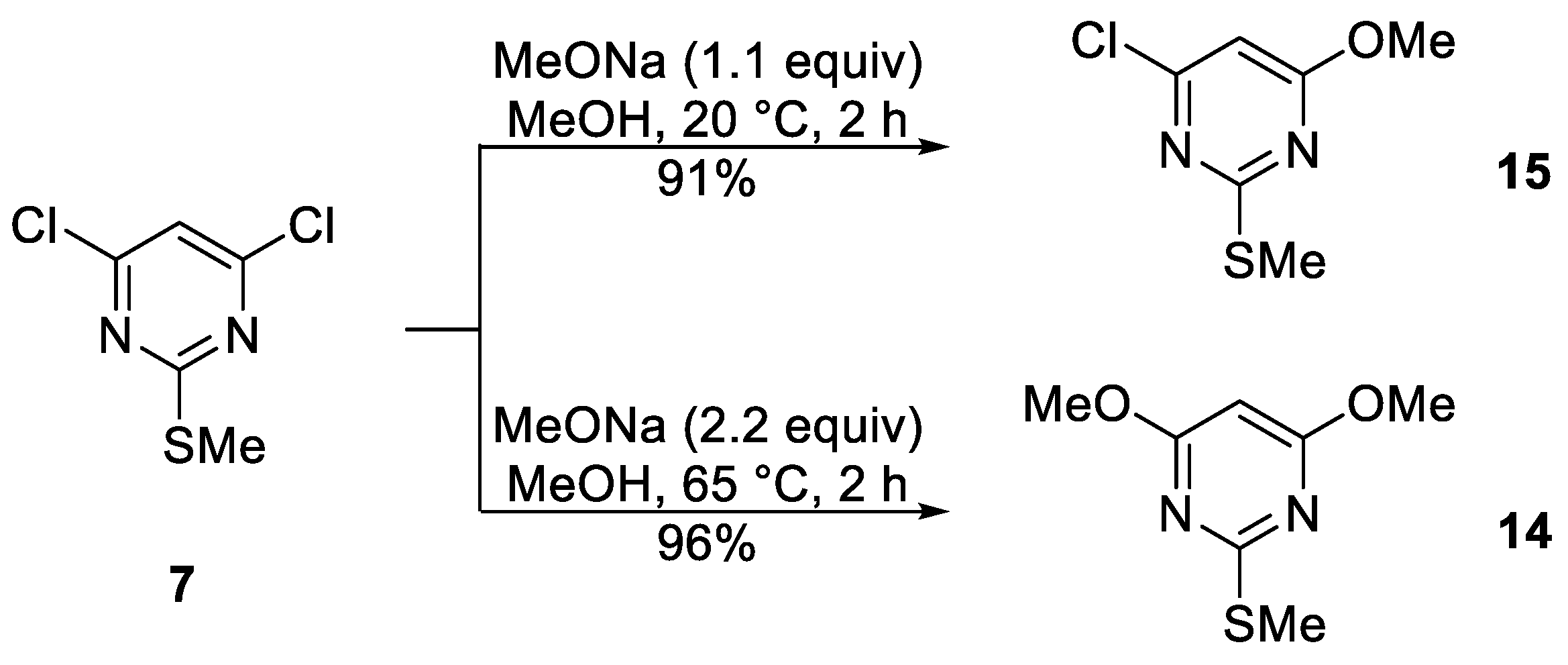
4-Chloro-6-methoxy-2-(methylthio)pyrimidine (15)
To a stirred mixture of 4,6-dichloro-2-(methylthio)pyrimidine (
7) (585 mg, 3.00 mmol) in MeOH (15 mL) at ca. 20 °C was added in one portion a solution of MeONa 1 M in MeOH (3.30 mL, 3.30 mmol). The mixture was protected with a CaCl
2 drying tube and stirred at this temperature until complete consumption of the starting material (TLC, 2 h). Et
2O (20 mL) and NaHCO
3 sat. (10 mL) were then added, the two layers separated, and the aqueous layer extracted with a further 10 mL of Et
2O. The combined organic phases were then dried over Na
2SO
4, filtered, and evaporated in vacuo to give the title compound
15 (518 mg, 91%) as a colorless oil; R
f 0.31 (
n-hexane/DCM, 70:30),
δH(500 MHz; CDCl
3) 6.41 (1H, s, C
H), 3.96 (3H, s, OC
H3), 2.55 (3H, s, SC
H3), identical to the one reported [
11].
4,6-Dimethoxy-2-(methylthio)pyrimidine (14)
To a stirred mixture of 4,6-dichloro-2-(methylthio)pyrimidine (
7) (780 mg, 4.00 mmol) in MeOH (15 mL) at ca. 20 °C was added in one portion a solution of MeONa 3 M in MeOH (2.93 mL, 8.80 mmol). The mixture was protected with a CaCl
2 drying tube and heated to ca. 65 °C until complete consumption of the starting material (TLC, 2 h). DCM (10 mL) was then added, the mixture adsorbed onto silica, and chromatography (
n-hexane/DCM 70:30) gave the title compound
14 (717 mg, 96%) as yellow plates, mp 53–54 °C (from
n-hexane/−40 °C, lit. 53–54 °C [
13]); R
f 0.18 (
n-hexane/DCM, 70:30);
δH(500 MHz; CDCl
3) 5.71 (1H, s, C
H), 3.92 (6H, s, OC
H3), 2.54 (6H, s, SC
H3), identical to the one reported [
14].
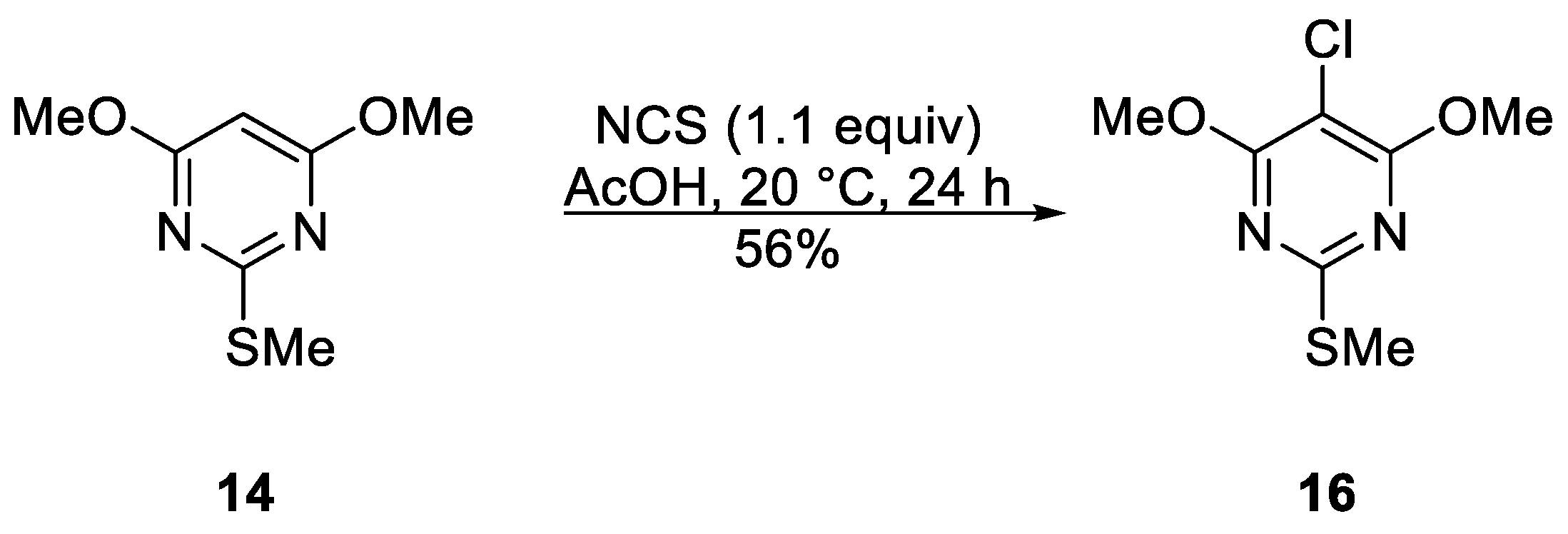
5-Chloro-4,6-dimethoxy-2-(methylthio)pyrimidine (16)
To a stirred mixture of 4,6-dimethoxy-2-(methylthio)pyrimidine (14) (93.1 mg, 0.500 mmol) in AcOH (1 mL) at ca. 20 °C was added in one portion N-chlorosuccinimide (73.4 mg, 0.55 mmol). The mixture was protected with a CaCl2 drying tube and stirred at this temperature until complete consumption of the starting material (TLC, 24 h). DCM (10 mL) was then added, the mixture adsorbed onto silica, and chromatography (n-hexane/DCM 70:30) gave the title compound 16 (61.6 mg, 56%) as colorless needles, mp 128–129 °C (from n-hexane/−40 °C); Rf 0.46 (n-hexane/DCM, 70:30); (found: C, 37.94; H, 4.12; N, 12.53. C7H9ClN2O2S requires C, 38.10; H, 4.11; N, 12.69%); λmax(DCM)/nm 258 inf (log ε 4.11), 267 (4.16); vmax/cm−1 2963w and 2930w (C-H), 1560m, 1555s, 1493m, 1458w, 1389m, 1358s, 1335w, 1319m, 1290m, 1269m, 1186m, 1182m, 1126s, 1070m, 926m, 770m; δH(500 MHz; CDCl3) 4.03 (6H, s, OCH3), 2.53 (3H, s, SCH3); δC(125 MHz; CDCl3) 167.5 (Cq), 165.1 (Cq), 95.2 (Cq), 55.0 (CH3), 14.4 (CH3); m/z (MALDI-TOF) 221 (M+-H + 2, 33%), 220 (M+, 75), 219 (M+ − H, 100), 206 (12).
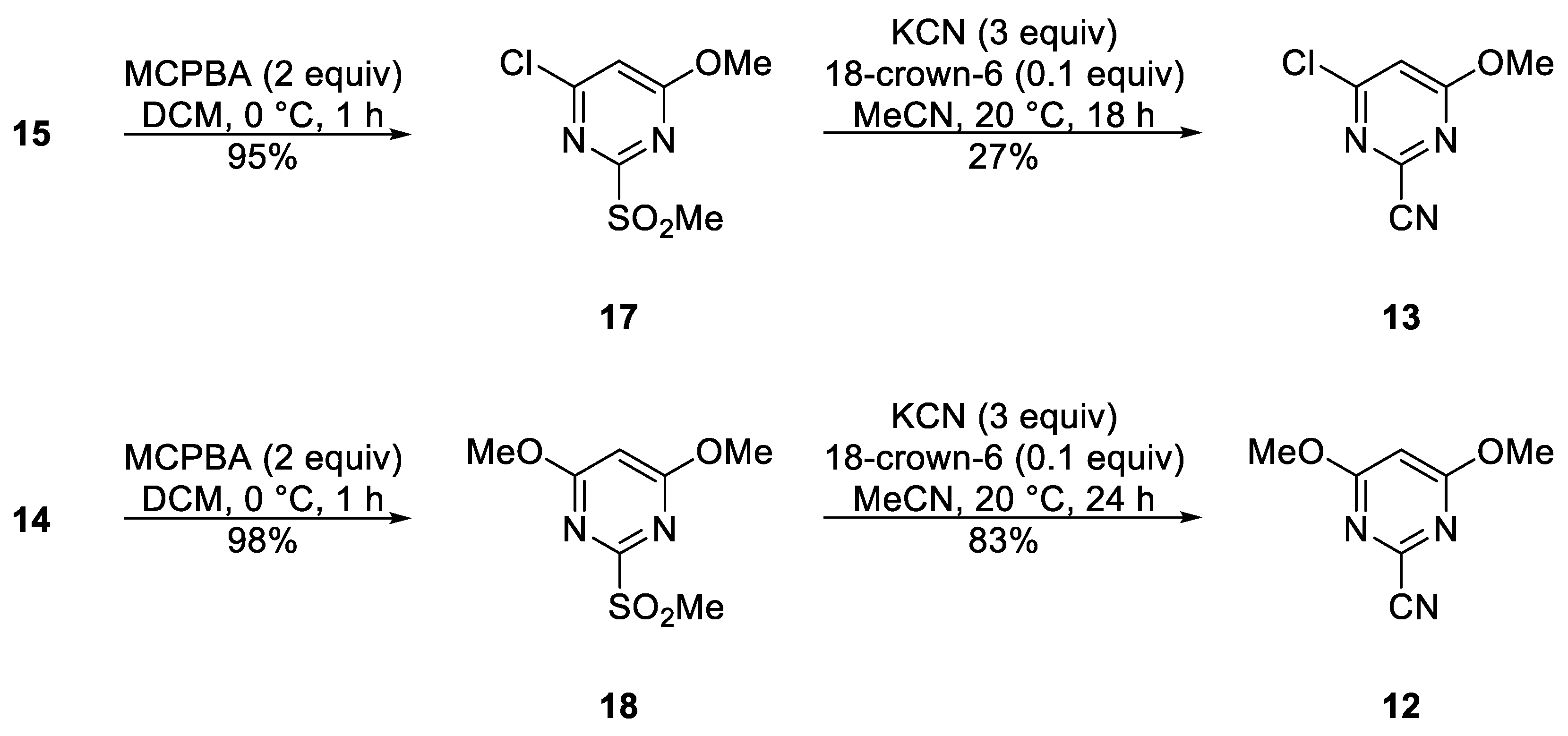
4-Chloro-6-methoxy-2-(methylsulfonyl)pyrimidine (17)
To a stirred mixture of 4-chloro-6-methoxy-2-(methylthio)pyrimidine (
15) (572 mg, 3.00 mmol) in DCM (10 mL) cooled in an ice-bath to ca. 0 °C was added in one portion m-chloroperbenzoic acid of 77% purity (1.344 g, 6.000 mmol). The mixture was protected with a CaCl
2 drying tube and stirred at this temperature until complete consumption of the starting material (TLC, 1 h). Et
2O (20 mL) and Na
2CO
3 sat. (10 mL) were then added, the two layers separated, and the aqueous layer extracted with a further 10 mL of Et
2O. The combined organic phases were then dried over Na
2SO
4, filtered, and evaporated in vacuo to give the title compound
17 (634 mg, 95%) as colorless needles, mp 85–86 °C (from
n-hexane/−40 °C, lit. 87–88 °C [
11]); R
f 0.22 (
n-hexane/DCM, 20:80);
δH(500 MHz; CDCl
3) 6.92 (1H, s, C
H), 4.10 (3H, s, OC
H3), 3.33 (3H, s, SO
2C
H3); identical to the one reported [
11].
4,6-Dimethoxy-2-(methylsulfonyl)pyrimidine (18)
To a stirred mixture of 4,6-dimethoxy-2-(methylthio)pyrimidine (
14) (186 mg, 1.00 mmol) in DCM (5 mL) cooled in an ice-bath to ca. 0 °C was added in one portion m-chloroperbenzoic acid of 77% purity (448 g, 3.000 mmol). The mixture was protected with a CaCl
2 drying tube and stirred at this temperature until complete consumption of the starting material (TLC, 1 h). Et
2O (20 mL) and Na
2CO
3 sat. (10 mL) were then added, the two layers separated, and the aqueous layer extracted with a further 10 mL of Et
2O. The combined organic phases were then dried over Na
2SO
4, filtered and evaporated in vacuo to give the title compound
18 (124 mg, 98%) as colorless plates, mp 125–126 °C (from EtOH, lit. 126–128 °C [
13]); R
f 0.74 (DCM);
δH(500 MHz; CDCl
3) 6.18 (1H, s, C
H), 4.03 (6H, s, OC
H3), 3.32 (3H, s, SO
2C
H3); identical to the one reported [
13].
4-Chloro-6-methoxypyrimidine-2-carbonitrile (13)
To a stirred mixture of 4-chloro-6-methoxy-2-(methylsulfonyl)pyrimidine (17) (223 mg, 1.00 mmol) in MeCN (5 mL) at ca. 20 °C was added in one portion 18-crown-6 (26 mg, 0.10 mmol) followed by KCN (195 mg, 3.00 mmol). The mixture was protected with a CaCl2 drying tube and stirred at this temperature until complete consumption of the starting material (TLC, 18 h). Et2O (20 mL) and H2O (10 mL) were then added, the two layers separated, and the aqueous layer extracted with a further 10 mL of Et2O. The combined organic phases were then dried over Na2SO4, filtered, and the mixture adsorbed onto silica and chromatography (n-hexane/DCM 20:80) gave the title compound 13 (48 mg, 27%) as colorless needles, mp 69–70 °C (from n-hexane/−40 °C); Rf 0.67 (n-hexane/DCM 20:80); (found: C, 42.65; H, 2.40; N, 24.63. C6H4ClN3O requires C, 42.50; H, 2.38; N, 24.78%); λmax(DCM)/nm 241 (log ε 3.59), 262 (3.41); vmax/cm−1 3084w (C-H), 1562s, 1533m, 1518m, 1470m, 1396m, 1360s, 1341m, 1244w, 1190m, 1130s, 1038s, 978s, 945m, 880m, 862m, 770w; δH(500 MHz; CDCl3) 6.95 (1H, s, CH), 4.06 (3H, s, OCH3); δC(125 MHz; CDCl3) 170.8 (Cq), 161.3 (Cq), 143.5 (Cq), 114.6 (Cq), 111.1 (CH), 55.7 (CH3); m/z (MALDI-TOF) 170 (M+-H + 2, 20%), 169 (M+, 40), 168 (M+ − H, 100), 132 (36).
4,6-Dimethoxypyrimidine-2-carbonitrile (12)
To a stirred mixture of 4,6-dimethoxy-2-(methylsulfonyl)pyrimidine (18) (218 mg, 1.00 mmol) in MeCN (5 mL) at ca. 20 °C was added in one portion 18-crown-6 (26 mg, 0.10 mmol) followed by KCN (195 mg, 3.00 mmol). The mixture was protected with a CaCl2 drying tube and stirred at this temperature until complete consumption of the starting material (TLC, 24 h). Et2O (20 mL) and H2O (10 mL) were then added, the two layers separated, and the aqueous layer extracted with a further 10 mL of Et2O. The combined organic phases were then dried over Na2SO4, filtered, and the mixture adsorbed onto silica and chromatography (n-hexane/DCM 60:40) gave the title compound 12 (138 mg, 83%) as colorless needles, mp 113–114 °C (from n-hexane/−40 °C); Rf 0.31 (n-hexane/DCM, 60:40); (found: C, 50.77; H, 4.35; N, 25.26. C7H7N3O2 requires C, 50.91; H, 4.27; N, 25.44%); λmax(DCM)/nm 259 (log ε 3.69); vmax/cm−1 3094w, 3003w, 2926w and 2849w (C-H), 1591s, 1566m, 1530m, 1468m, 1387m, 1352m, 1202s, 1173m, 1059s, 984m, 964m, 864m, 772m; δH(500 MHz; CDCl3) 6.20 (1H, s, CH), 3.98 (6H, s, OCH3); δC(125 MHz; CDCl3) 171.2 (Cq), 142.6 (Cq), 115.5 (Cq), 94.1 (CH), 54.9 (CH3); m/z (MALDI-TOF) 166 (MH+, 100%), 165 (M+, 25), 164 (M+ − H, 51), 162 (55).
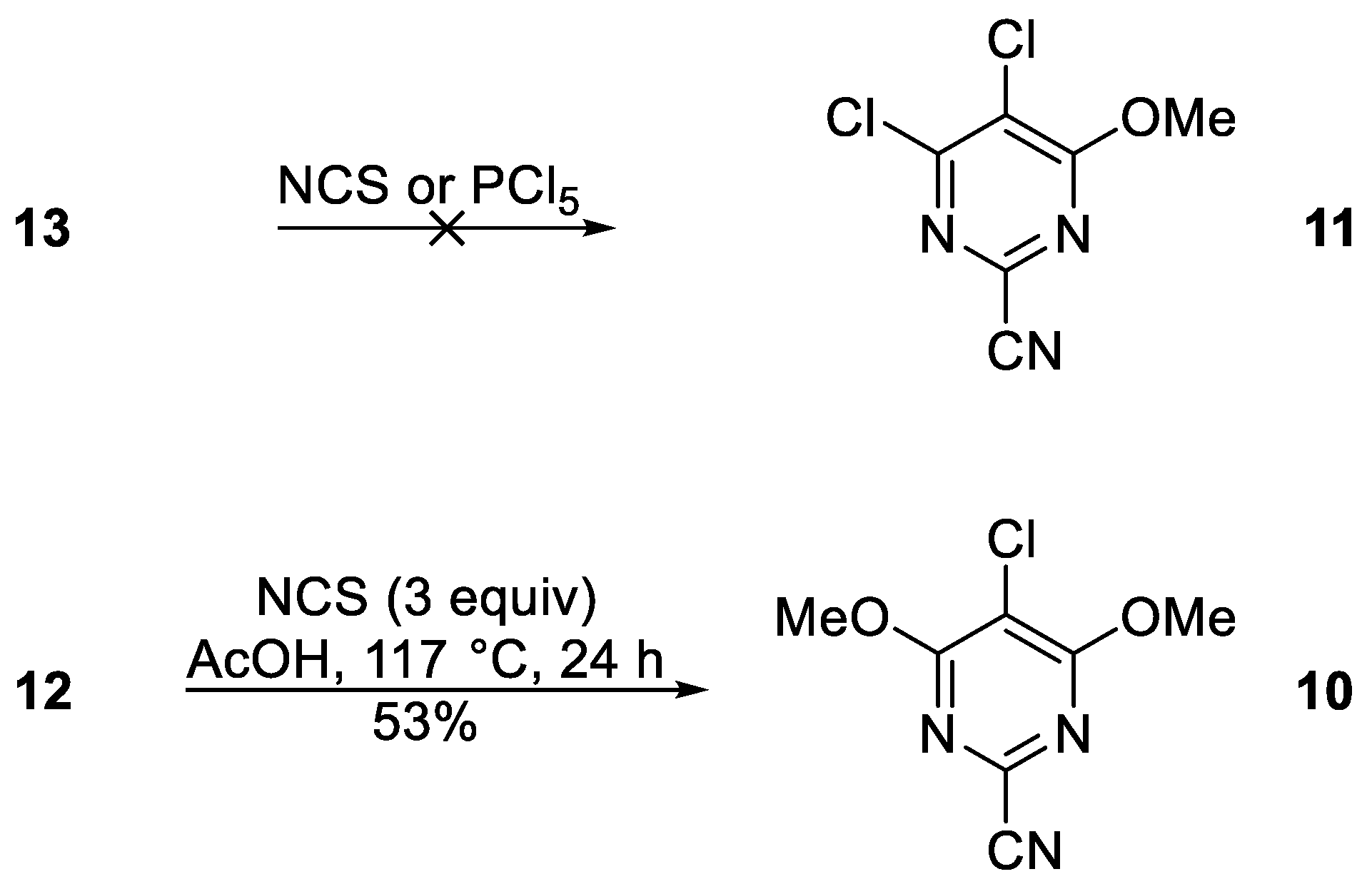
5-Chloro-4,6-dimethoxypyrimidine-2-carbonitrile (10)
To a stirred mixture of 4,6-dimethoxypyrimidine-2-carbonitrile (12) (50.0 mg, 0.303 mmol) in AcOH (2 mL) at ca. 20 °C was added in one portion N-chlorosuccinimide (121 mg, 0.908 mmol). The mixture was protected with a CaCl2 drying tube and stirred at ca. 117 °C until complete consumption of the starting material (TLC, 24 h). Et2O (20 mL) and H2O (10 mL) were then added, the two layers separated, and the aqueous layer extracted with a further 10 mL of Et2O. The combined organic phases were then dried over Na2SO4, filtered, and the mixture adsorbed onto silica and chromatography (n-hexane/DCM 60:40) gave the title compound 10 (31.8 mg, 53%) as colorless needles, mp 145–147 °C (from n-hexane/−40 °C); Rf 0.48 (n-hexane/DCM, 60:40); (found: C, 41.96; H, 2.89; N, 20.96. C7H6ClN3O2 requires C, 42.12; H, 3.03; N, 21.05%); λmax(DCM)/nm 278 (log ε 3.66), 290 (3.45); vmax/cm−1 3003w, 2980w and 2918w (C-H), 1572s, 1566s, 1545w, 1524w, 1493w, 1462m, 1443w, 1385m, 1373s, 1362m, 1294m, 1182m, 1126s, 1088w, 1076w, 1013w, 968m, 914m, 781s, 741w, 712m; δH(500 MHz; CDCl3) 4.09 (6H, s, OCH3); δC(125 MHz; CDCl3) 166.0 (Cq), 138.5 (Cq), 115.1 (Cq), 104.4 (Cq), 56.1 (CH3); m/z (MALDI-TOF) 202 (MH+ + 2, 34%), 201 (M+ + 2,29), 200 (M+,38), 199 (M+, 100), 192 (41), 156 (10).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





