4b,5,6,9-Tetrahydro-7H-dibenzo[c,e]pyrrolo[1,2-a]azepin-7-one

 and
and 
Abstract
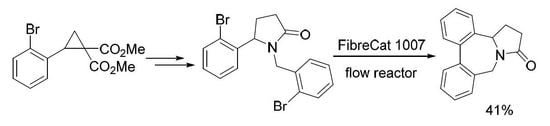
{kind=link}
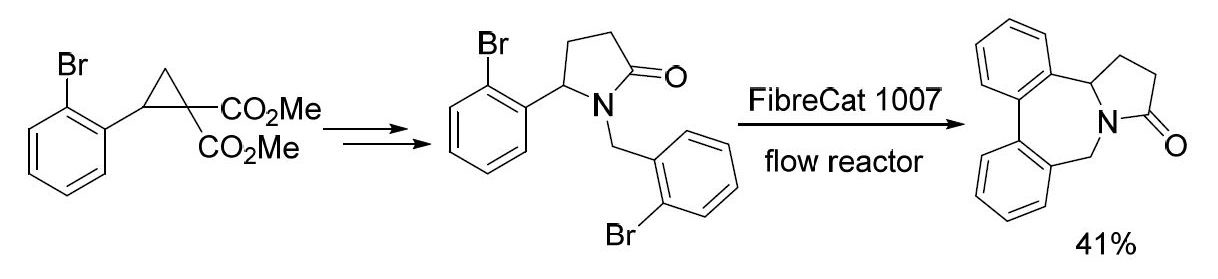
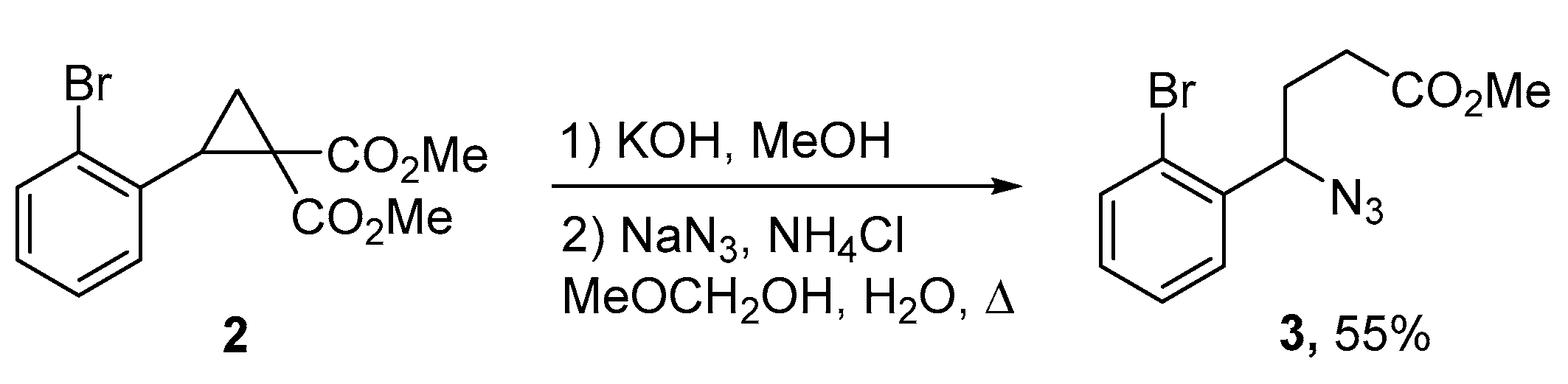{kind=link}
{kind=link}
{kind=link}
1. Introduction
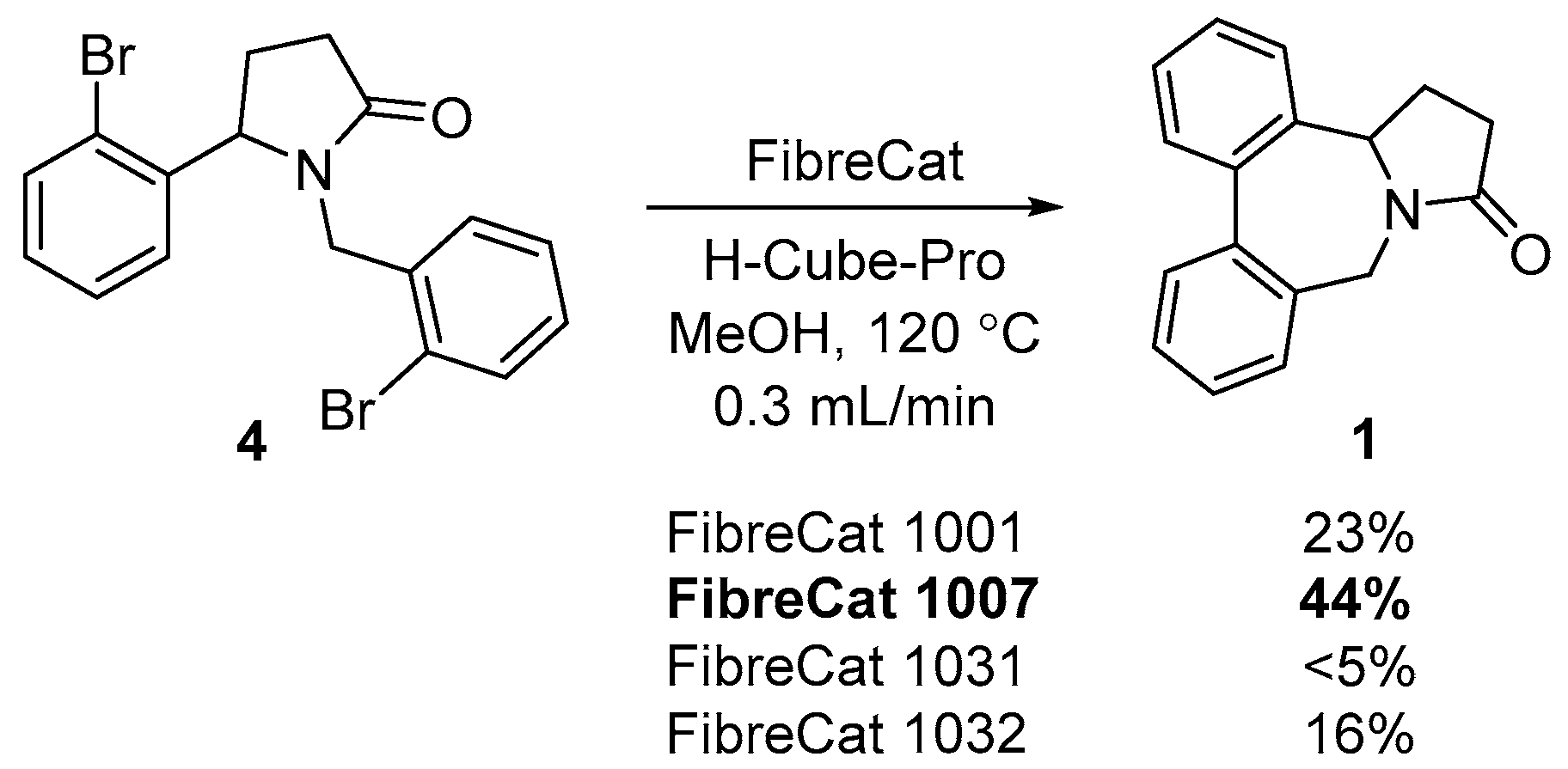
2. Results and Discussion
3. Materials and Methods
3.1. Methyl 4-Azido-4-(2-bromophenyl)butanoate (3)
3.2. 1-(2-Bromobenzyl)-5-(2-bromophenyl)pyrrolidin-2-one (4)
Continuous Flow Procedure
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stallworth, J.M.; Jeffords, J.V. Clinical Effects of Azapetine (Ilidar) on Peripheral Arterial Disease. JAMA 1956, 161, 840–843. [Google Scholar] [CrossRef]
- Qiu, J.; Chen, W.; Jiang, Y.; Chen, J.; Zhang, Y.; Gu, X. Assessment of a bifendate derivative bearing a 6,7-dihydro-dibenzo[c,e]azepine scaffold as a potential anti-metastatic agent. Med. Chem. Commun. 2018, 9, 1826–1830. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Jiang, Y.; Qu, Y.; Chen, J.; Feng, D.; Li, C.; Yin, X. Synthesis and biological evaluation of bifendate derivatives bearing 6,7-dihydro-dibenzo[c,e]azepine scaffold as potential P-glucoprotein and tumor metastasis inhibitors. Eur. J. Med. Chem. 2018, 145, 379–388. [Google Scholar] [CrossRef]
- Gu, X.; Tang, X.; Zhao, Q.; Peng, H.; Peng, S.; Zhang, Y. Discovery of alkoxy biphenyl derivatives bearing dibenzo[c,e]azepine scaffold as potential dual inhibitors of P-glycoprotein and breast cancer resistance protein. Bioorg. Med. Chem. Lett. 2014, 24, 3419–3421. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Ren, Z.; Tang, X.; Peng, H.; Zhao, Q.; Lai, Y.; Peng, S.; Zhang, Y. Synthesis and biological evaluation of novel bifendate derivatives bearing 6,7-dihydro-dibenzo[c,e]azepine scaffold as potent P-glycoprotein inhibitors. Eur. J. Med. Chem. 2012, 51, 137–144. [Google Scholar] [CrossRef]
- Mehta, V.P.; Modha, S.G.; Ruijter, E.; Van Hecke, K.; Van Meervelt, L.; Pannecouque, C.; Balzarini, J.; Orru, R.V.A.; Van der Eycken, E. A Microwave-Assisted Diastereoselective Multicomponent Reaction To Access Dibenzo[c,e]azepinones: Synthesis and Biological Evaluation. J. Org. Chem. 2011, 76, 2828–2839. [Google Scholar] [CrossRef]
- Edwards, D.J.; Hadfield, J.A.; Wallace, T.W.; Ducki, S. Tubulin-binding dibenz[c,e]oxepines as colchicinol analogues for targeting tumour vasculature. Org. Biomol. Chem. 2011, 9, 219–231. [Google Scholar] [CrossRef]
- Hall, I.H.; Barnes, B.J.; Ward, E.S.; Wheaton, J.R.; Shaffer, K.A.; Cho, S.E.; Warren, A.E. Targeting of Human Tmolt4 Leukemic Type II IMP Dehydrogenase by Cyclic Imide Related Derivatives. Arch. Pharm. Pharm. Med. Chem. 2001, 334, 229–234. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.M.; ElTahir, K.E.H.; Asiri, Y.A. Synthesis, anti-inflammatory activity and COX-1/COX-2 inhibition of novel substituted cyclic imides. Part 1: Molecular docking study. Eur. J. Med. Chem. 2011, 46, 1648–1655. [Google Scholar] [CrossRef]
- Hall, I.H.; Wong, O.T.; Reynolds, D.J.; Simlot, R. Comparison between 6,7-Dihydro-5H-Dibenz(c,e)-azepine and Lovastatin as Hypolipidemic Agents in Rats. J. Pharm. Sci. 1993, 82, 565–570. [Google Scholar] [CrossRef]
- Hall, I.H.; Murthy, A.R.K.; Wyrick, S.D. Hypolipidemic Activity of 6-Substituted 6,7-Dihydro-5H-Dibenz(c,e)azepine and the Effects of 6,7-Dihydro-5H-Dibenz(c,e)azepine on Lipid Metabolism of Rodents. J. Pharm. Sci. 1986, 75, 622–626. [Google Scholar] [CrossRef]
- De Lera Ruiz, M.; Zheng, J.; Berlin, B.Y.; McCormick, K.D.; Aslanian, R.G.; West, R.; Hwa, J.; Lachowicz, J.; van Heek, M. Bicyclic and tricyclic heterocycle derivatives as histamine H3 receptor antagonists for the treatment of obesity. Bioorg. Med. Chem. Lett. 2013, 23, 6004–6009. [Google Scholar] [CrossRef]
- Tang, X.; Gu, X.; Ren, Z.; Ma, Y.; Lai, Y.; Peng, H.; Peng, S.; Zhang, Y. Synthesis and evaluation of substituted dibenzo[c,e]azepin-5-ones as P-glycoprotein-mediated multidrug resistance reversal agents. Bioorg. Med. Chem. Lett. 2012, 22, 2675–2680. [Google Scholar] [CrossRef] [PubMed]
- Hadden, M.; Goodman, A.; Guo, C.; Guzzo, P.R.; Henderson, A.J.; Pattamana, K.; Ruenz, M.; Sargent, B.J.; Swenson, B.; Yet, L.; et al. Synthesis and SAR of heterocyclic carboxylic acid isosteres based on 2-biarylethylimidazole as bombesin receptor subtype-3 (BRS-3) agonists for the treatment of obesity. Bioorg. Med. Chem. Lett. 2010, 20, 2912–2915. [Google Scholar] [CrossRef]
- Schmidt, M.; Teitge, M.; Castillo, M.E.; Brandt, T.; Dobner, B.; Langner, A. Synthesis and Biochemical Characterization of New Phenothiazines and Related Drugs as MDR Reversal Agents. Arch. Pharm. Chem. Life Sci. 2008, 341, 624–638. [Google Scholar] [CrossRef]
- Büttner, F.; Bergemann, S.; Guenard, D.; Gust, R.; Seitz, G.; Thoret, S. Two novel series of allocolchicinoids with modified seven membered B-rings: Design, synthesis, inhibition of tubulin assembly and cytotoxicity. Bioorg. Med. Chem. 2005, 13, 3497–3511. [Google Scholar] [CrossRef]
- Reynolds, D.J.; Wong, O.T.; Simlot, R.; Chang, J.J.; Hall, I.H. Acute Toxic and Teratogenic Effects of Cyclic Imides in Rodent. Arch. Pharm. 1994, 327, 237–245. [Google Scholar] [CrossRef]
- Gorshkova, V.K.; Saratikov, A.S.; Tignibidina, L.G. Investigation of the Anticonvulsant and Antihypoxic Activity of New Dibenzazepine Derivatives. Pharm. Chem. J. 1994, 28, 158–162. [Google Scholar] [CrossRef]
- Wang, Y.G.; Ueda, M.; Wang, X.; Han, Z.; Maruoka, K. Convenient preparation of chiral phase-transfer catalysts with conformationally fixed biphenyl core for catalytic asymmetric synthesis of α-alkyl- and α,α-dialkyl-α-amino acids: Application to the short asymmetric synthesis of BIRT-377. Tetrahedron 2007, 63, 6042–6050. [Google Scholar] [CrossRef]
- Kan, S.B.J.; Maruyama, H.; Akakura, M.; Kano, T.; Maruoka, K. Catalyst-Controlled, Enantioselective and Diastereodivergent Conjugate Addition of Aldehydes to Electron-Deficient Olefins. Angew. Chem. Int. Ed. 2017, 56, 9487–9491. [Google Scholar] [CrossRef] [PubMed]
- Boichenko, M.A.; Chagarovskiy, A.O. Recent achievements in the synthesis of dibenz[c,e]azepines. Chem. Heterocycl. Comp. 2017, 53, 1280–1282. [Google Scholar] [CrossRef]
- Balgobin, S.M.C.; Brookes, D.J.; Jiang, J.; Pritchard, R.G.; Wallace, T.W. Axial stereocontrol in tropos dibenz[c,e]azepines: The individual and cooperative effects of alkyl substituents. Org. Biomol. Chem. 2017, 15, 10184–10199. [Google Scholar] [CrossRef]
- Page, P.C.B.; Pearce, C.A.; Chan, Y.; Parker, P.; Buckley, B.R.; Rassias, G.A.; Elsegood, M.R.J. Atropo- and Diastereoselective Construction of Tetracyclic Biphenylazepinium Salts Derived from Aminoalcohols: Use as Catalysts in Enantioselective Asymmetric Epoxidation. J. Org. Chem. 2015, 80, 8036–8045. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhao, L.; Li, Y.; Soule, J.F.; Doucet, H. Intermolecular versus Intramolecular Palladium-Catalyzed Direct Arylations between 1-(2-Bromoimidazoles) and Aryl Bromides. Adv. Synth. Catal. 2015, 357, 2869–2882. [Google Scholar] [CrossRef]
- Cheetham, C.A.; Massey, R.S.; Pira, S.L.; Pritchard, R.G.; Wallace, T.W. Atroposelective formation of dibenz[c,e]azepines via intramolecular direct arylation with centre-axis chirality transfer. Org. Biomol. Chem. 2011, 9, 1831–1838. [Google Scholar] [CrossRef]
- Yu, M.; Tang, R.Y.; Li, J.H. Synthesis of 6,7-dihydro-5H-dibenzo[c,e]azepines and biaryls by palladium-catalyzed Ullmann reaction. Tetrahedron 2009, 65, 3409–3416. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Mondal, S.; De, N. Synthesis of Polycyclic Sultams by Palladium-Catalyzed Intramolecular Cyclization. Synthesis 2009, 3127–3135. [Google Scholar] [CrossRef]
- Saudan, L.A.; Bernardinelli, G.; Kündig, E.P. Diastereoselective synthesis of (5R,7R)- and (5R,7S)-5,7-Dimethyl-6,7-dihydro-5H-dibenz[c,e]azepines. Synlett 2000, 483–486. [Google Scholar] [CrossRef]
- Hennings, D.D.; Iwama, T.; Rawal, V.H. Palladium-Catalyzed (Ullmann-Type) Homocoupling of Aryl Halides: A Convenient and General Synthesis of Symmetrical Biaryls via Inter- and Intramolecular Coupling Reactions. Org. Lett. 1999, 1, 1205–1208. [Google Scholar] [CrossRef]
- Ivanov, K.L.; Villemson, E.V.; Budynina, E.M.; Ivanova, O.A.; Trushkov, I.V.; Melnikov, M.Y. Ring Opening of Donor-Acceptor Cyclopropanes with the Azide Ion: A Tool for Construction of N-Heterocycles. Chem. Eur. J. 2015, 21, 4975–4987. [Google Scholar] [CrossRef]
- Budynina, E.M.; Ivanov, K.L.; Chagarovskiy, A.O.; Rybakov, V.B.; Trushkov, I.V.; Melnikov, M. Ya. From Umpolung to Alternation: Modified Reactivity of Donor-Acceptor Cyclopropanes Towards Nucleophiles in Reaction with Nitroalkanes. Chem. Eur. J. 2016, 22, 3692–3696. [Google Scholar] [CrossRef]
- Pavlova, A.S.; Ivanova, O.A.; Chagarovskiy, A.O.; Stebunov, N.S.; Orlov, N.V.; Shumsky, A.N.; Budynina, E.M.; Rybakov, V.B.; Trushkov, I.V. Domino Staudinger/aza-Wittig/Mannich Reaction: An Approach to Diversity of Di- and Tetrahydropyrrole Scaffolds. Chem. Eur. J. 2016, 22, 17967–17971. [Google Scholar] [CrossRef] [PubMed]
- Villemson, E.V.; Budynina, E.M.; Ivanova, O.A.; Skvortsov, D.A.; Trushkov, I.V.; Melnikov, M. Ya. Concise Approach to pyrrolizino[1,2-b]indoles from indole-derived donor-acceptor cyclopropanes. RSC Adv. 2016, 6, 62014–62018. [Google Scholar] [CrossRef]
- Chagarovskiy, A.O.; Ivanova, O.A.; Shumsky, A.N.; Trushkov, I.V. Synthesis of hexahydropyridazin-3-ones by reaction between donor-acceptor cyclopropanes and phenylhydrazine. Chem. Heterocyclic Comp. 2017, 53, 1220–1227. [Google Scholar] [CrossRef]
- Boichenko, M.A.; Ivanova, O.A.; Andreev, I.A.; Chagarovskiy, A.O.; Levina, I.I.; Rybakov, V.B.; Skvortsov, D.A.; Trushkov, I.V. Convenient approach to polyoxygenated dibenzo[c,e]pyrrolo[1,2-a]azepines from donor-acceptor cyclopropanes. Org. Chem. Front. 2018, 5, 2829–2834. [Google Scholar] [CrossRef]
- Emmett, M.R.; Grover, H.K.; Kerr, M.A. Tandem Ring-Opening Decarboxylation of Cyclopropane Hemimalonates with Sodium Azide: A Short Route to γ-Aminobutyric Acid Esters. J. Org. Chem. 2012, 77, 6634–6637. [Google Scholar] [CrossRef]
- Hayashi, Y. Pot economy and one-pot synthesis. Chem. Sci. 2016, 7, 866–880. [Google Scholar] [CrossRef]
- Feiz, A.; Bazgir, A.; Balu, A.M.; Luque, R. Continuous flow room temperature reductive aqueous homo-coupling of aryl halides using supported Pd catalysts. Sci. Rep. 2016, 6, 32719. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boichenko, M.A.; Babkin, I.Y.; Kobylskoy, S.G.; Chagarovskiy, A.O.; Ivanova, O.A.; Trushkov, I.V. 4b,5,6,9-Tetrahydro-7H-dibenzo[c,e]pyrrolo[1,2-a]azepin-7-one. Molbank 2019, 2019, M1061. https://doi.org/10.3390/M1061
Boichenko MA, Babkin IY, Kobylskoy SG, Chagarovskiy AO, Ivanova OA, Trushkov IV. 4b,5,6,9-Tetrahydro-7H-dibenzo[c,e]pyrrolo[1,2-a]azepin-7-one. Molbank. 2019; 2019(2):M1061. https://doi.org/10.3390/M1061
Chicago/Turabian StyleBoichenko, Maksim A., Igor Yu. Babkin, Sergey G. Kobylskoy, Alexey O. Chagarovskiy, Olga A. Ivanova, and Igor V. Trushkov. 2019. "4b,5,6,9-Tetrahydro-7H-dibenzo[c,e]pyrrolo[1,2-a]azepin-7-one" Molbank 2019, no. 2: M1061. https://doi.org/10.3390/M1061
APA StyleBoichenko, M. A., Babkin, I. Y., Kobylskoy, S. G., Chagarovskiy, A. O., Ivanova, O. A., & Trushkov, I. V. (2019). 4b,5,6,9-Tetrahydro-7H-dibenzo[c,e]pyrrolo[1,2-a]azepin-7-one. Molbank, 2019(2), M1061. https://doi.org/10.3390/M1061