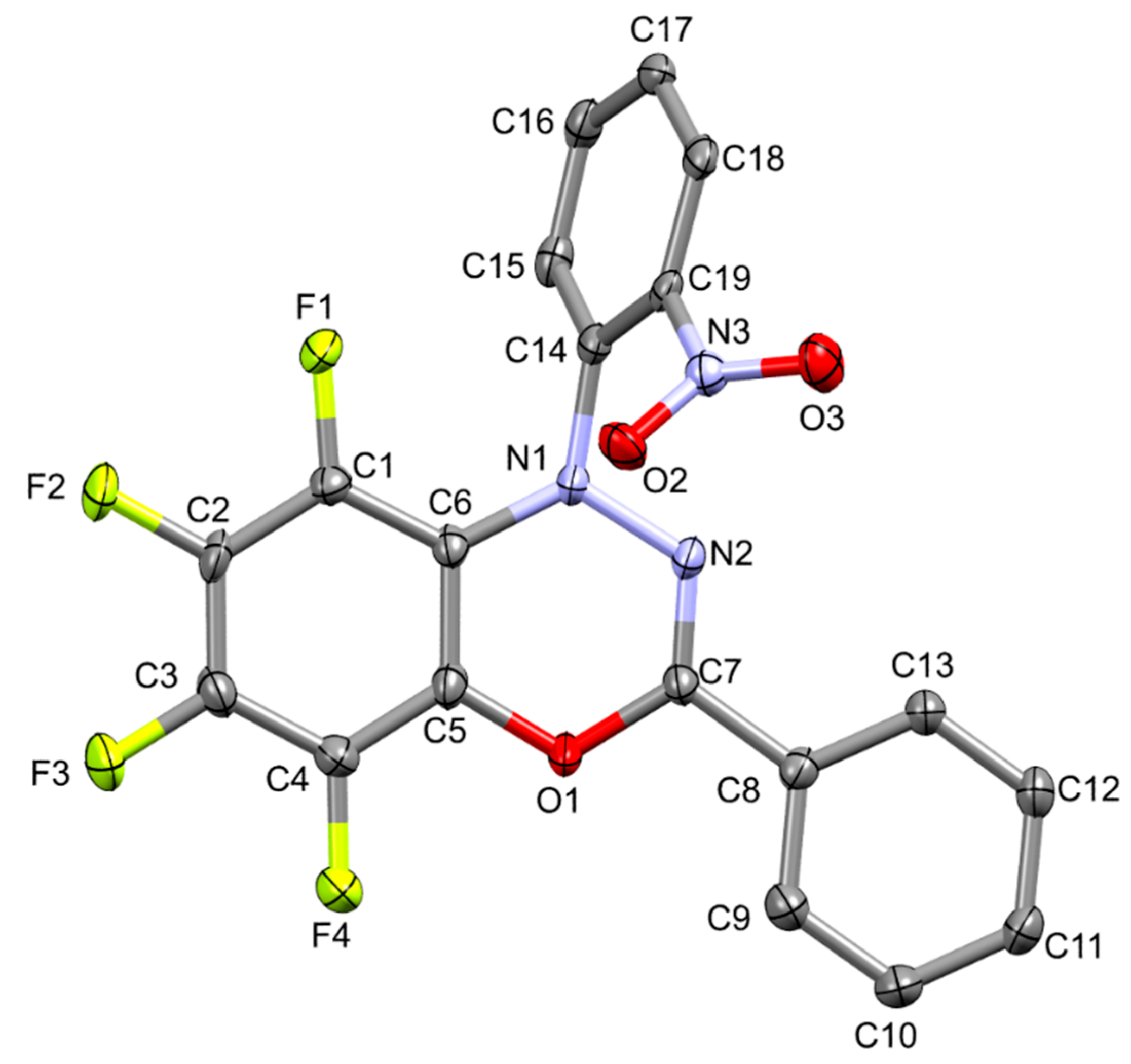
5,6,7,8-Tetrafluoro-1-(2-nitrophenyl)-3-phenyl-1H-benzo[e][1,3,4]oxadiazine



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
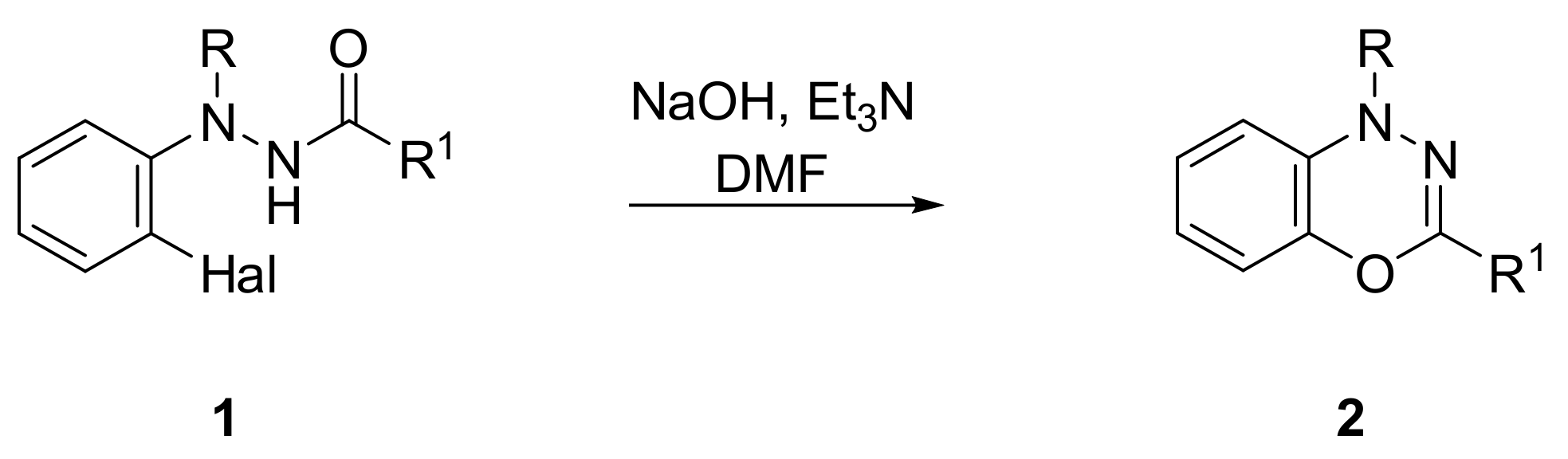
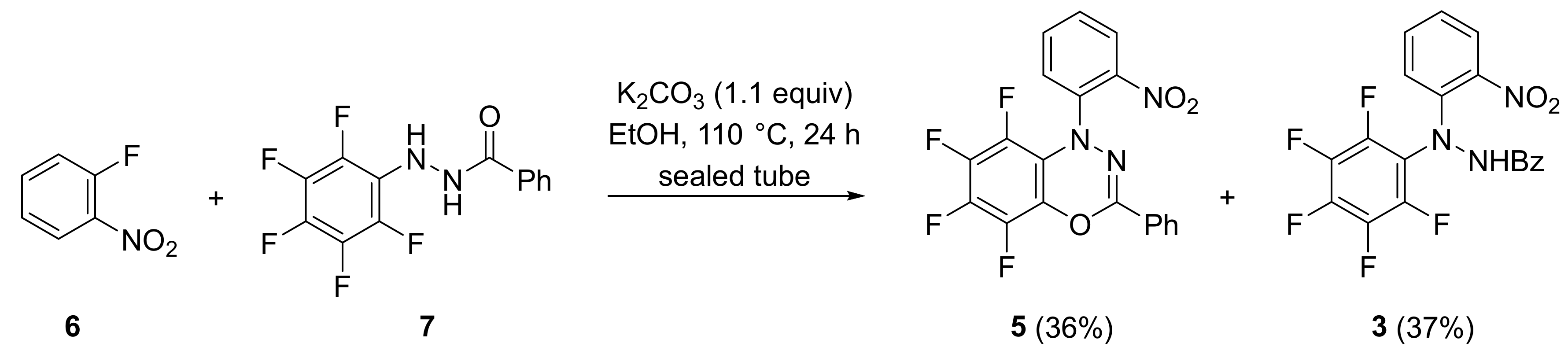
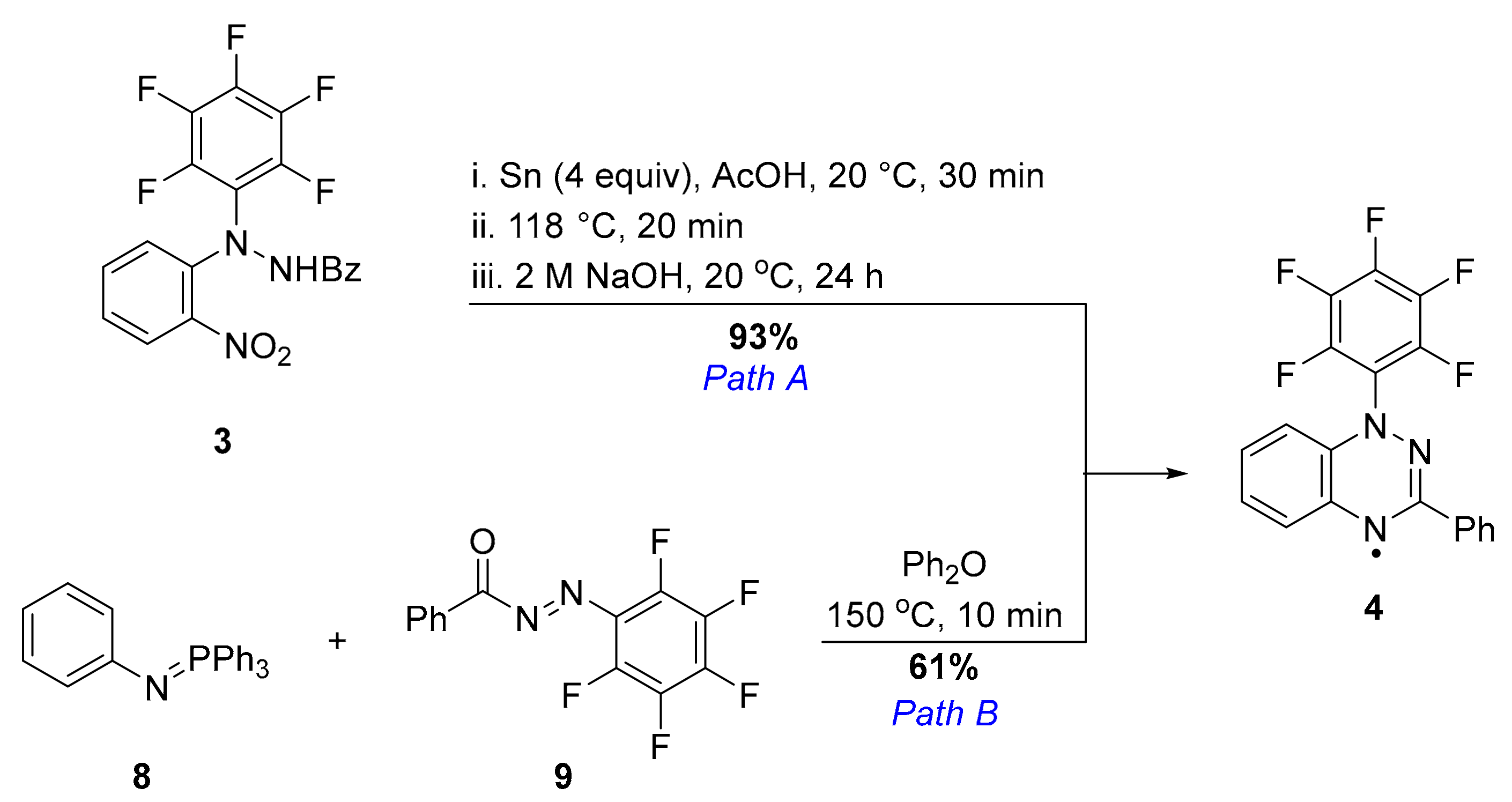
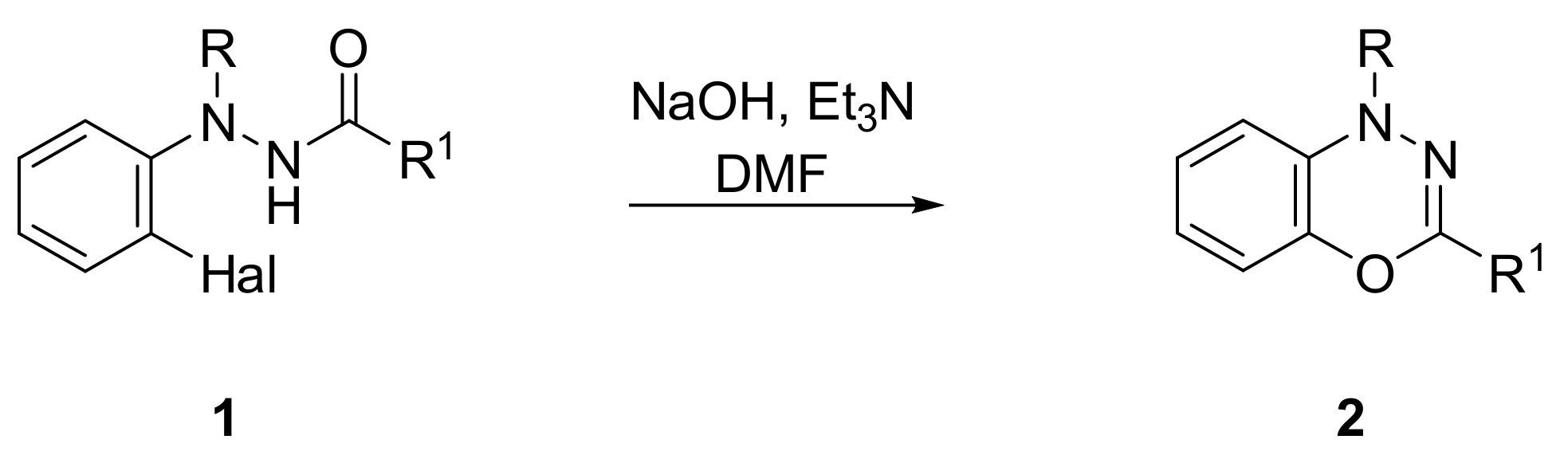
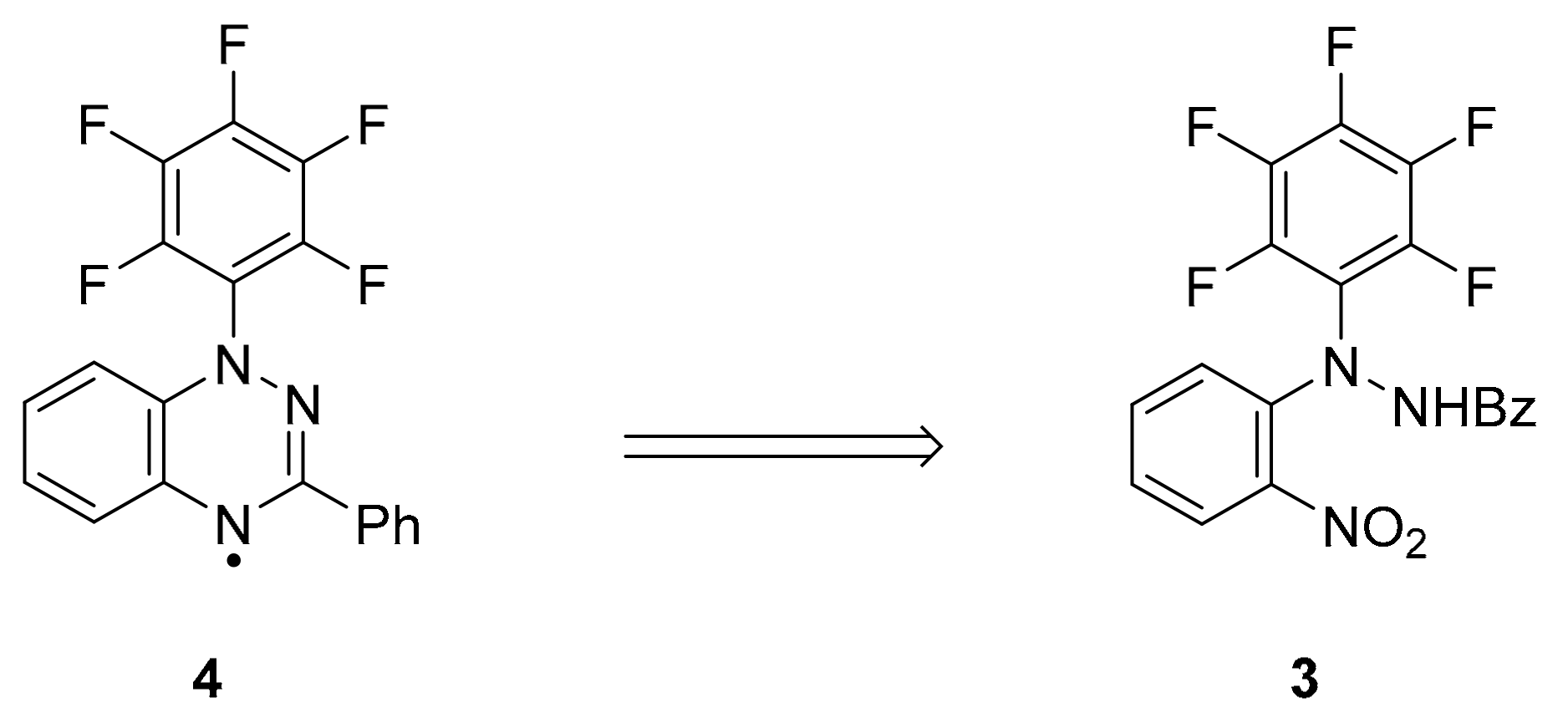
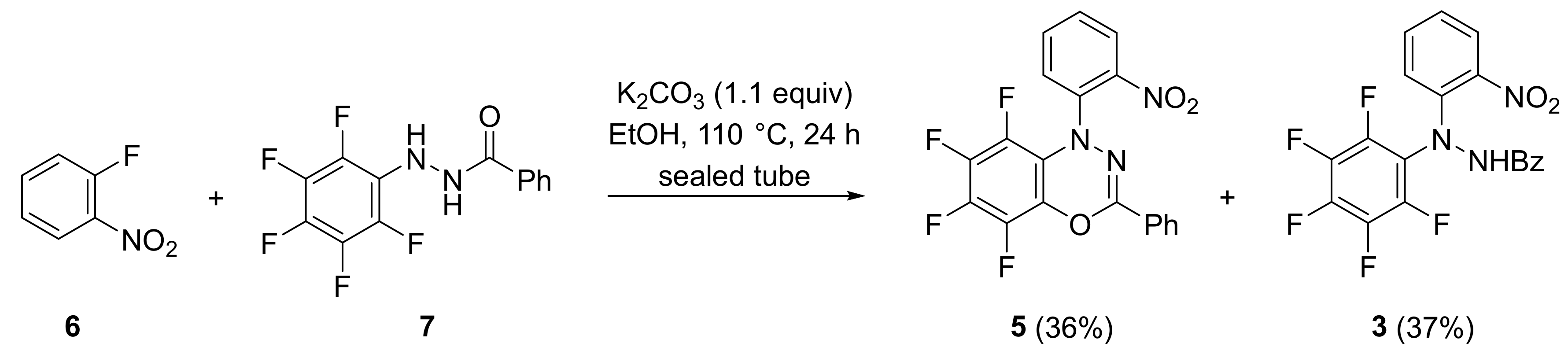
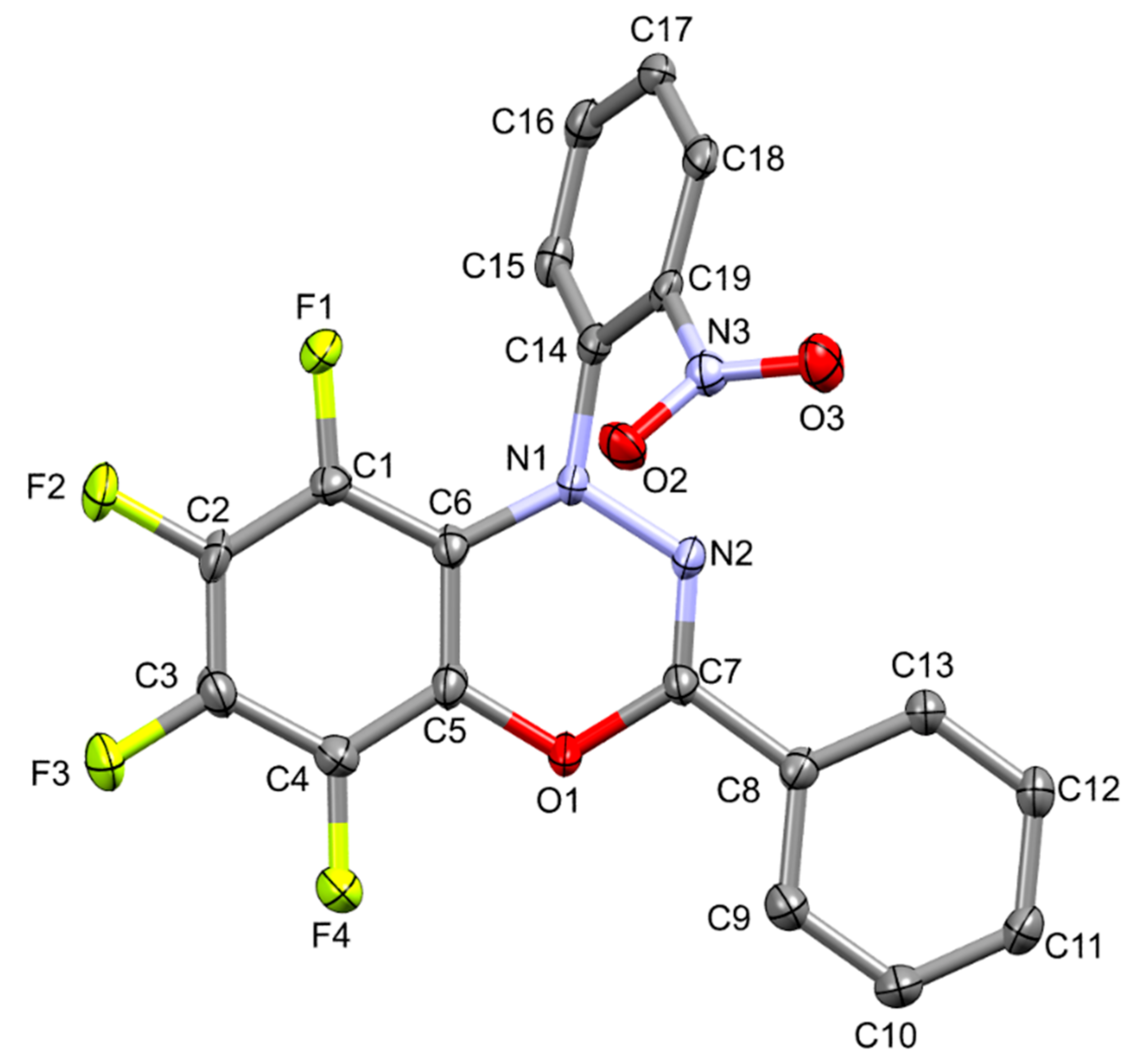
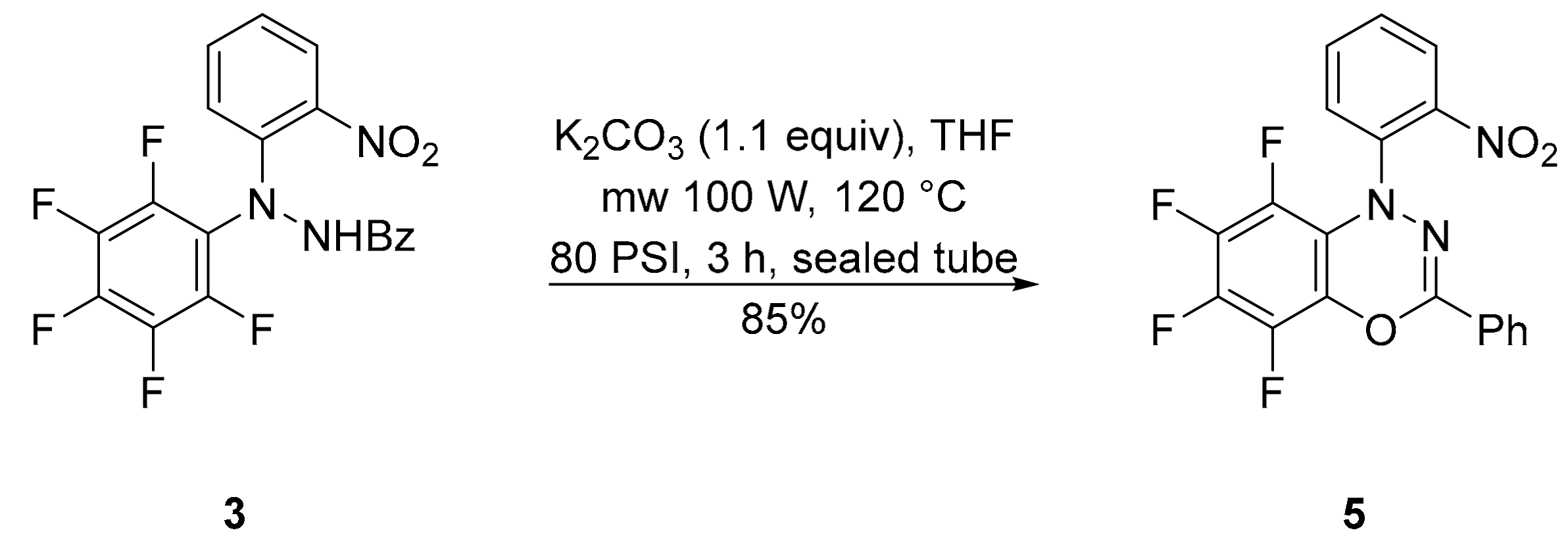
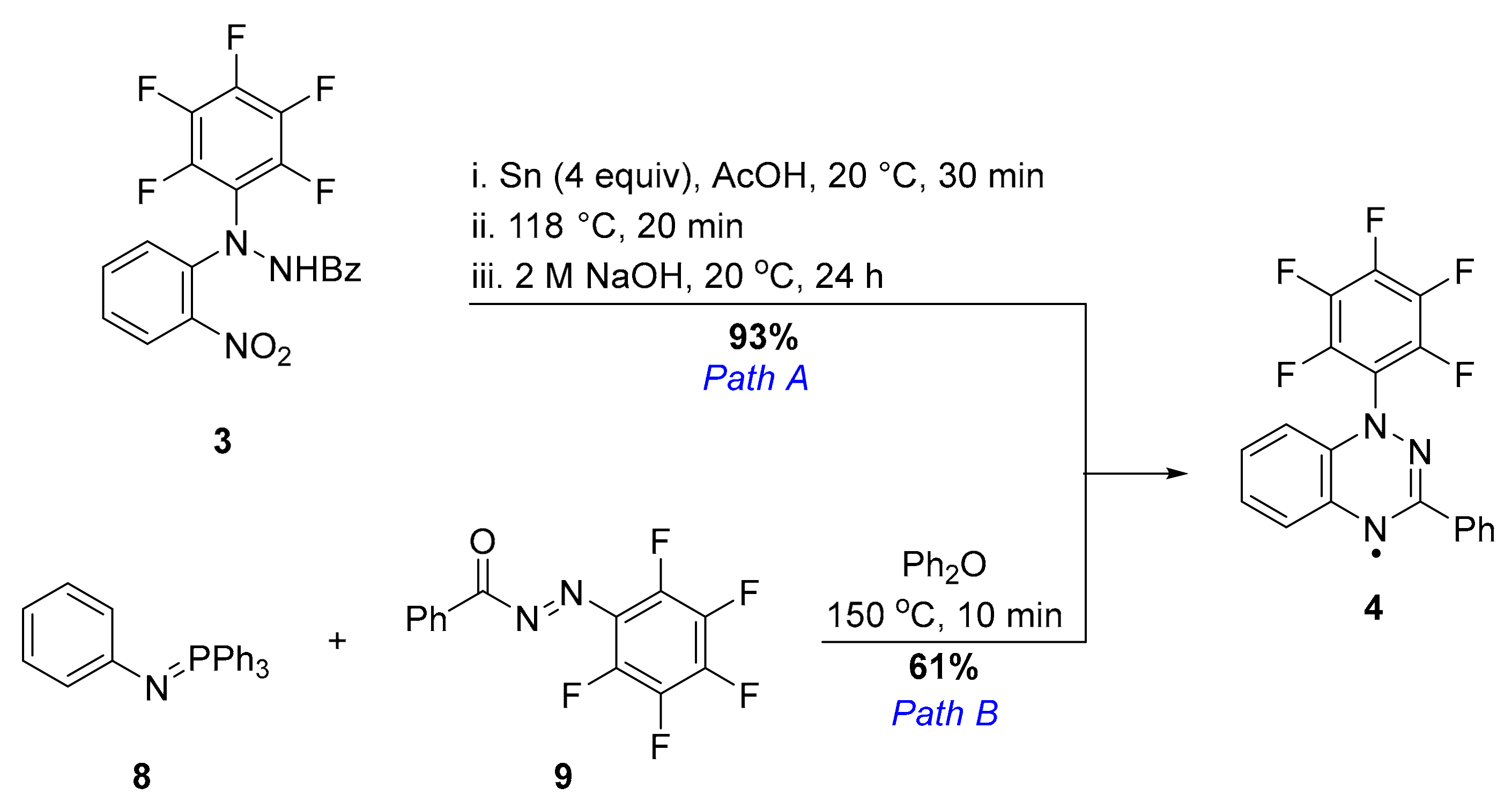
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. 5,6,7,8-Tetrafluoro-1-(2-nitrophenyl)-3-phenyl-1H-benzo[e][1,3,4]oxadiazine (5)
3.3. Conversion of N′-(2-Nitrophenyl)-N′-(perfluorophenyl)benzohydrazide (3) into 5,6,7,8-Tetrafluoro-1-(2-nitrophenyl)-3-phenyl-1H-benzo[e][1,3,4]oxadiazine (5)
3.4. 1-(Perfluorophenyl)-3-phenyl-1,2,4-benzotriazin-4-yl (4)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ke, S.; Cao, X.; Liang, Y.; Wang, K.; Yang, Z. Synthesis and Biological Properties of Dihydro-Oxadiazine-Based Heterocyclic Derivatives. Mini-Rev. Med. Chem. 2011, 11, 642–657. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, W.-D. 1,3,4-Oxadiazines and 1,3,4-Thiadiazines. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Pergamon Press: Oxford, UK, 2008. [Google Scholar]
- Eissa, F.M.; Abdelghany, A.R. New 1,3,4-Oxadiazinoisoquinoline Methine Cyanine Dyes: Synthesis, Photosensitivity and Antibacterial Activity. J. Heterocycl. Chem. 2016, 53, 429–436. [Google Scholar] [CrossRef]
- Eissa, F.M. Preparation, Antibacterial Activity and Absorption Spectra of Pyrazolo-Oxadiazine Cyanine Dyes. J. Chin. Chem. Soc. 2009, 56, 843–849. [Google Scholar] [CrossRef]
- Lipunova, G.N.; Nosova, É.V.; Sidorova, L.P.; Charushin, V.N. Synthesis and antitumor activity of fluorinated derivatives of [i,j]-annelated quinolones. Pharm. Chem. J. 2011, 45, 208–210. [Google Scholar] [CrossRef]
- Mousavi, S.-H.; Atapour-Mashhad, H.; Bakavoli, M.; Shiri, A.; Akbarzadeh, M.; Tayarani-Najaran, Z. Pyrimidooxadiazine and triazolopyrimidooxadiazine derivatives: Synthesis and cytotoxic evaluation in human cancer cell lines. Russ. J. Bioorg. Chem. 2015, 41, 201–208. [Google Scholar] [CrossRef]
- Bakavoli, M.; Rahimizadeh, M.; Shiri, A.; Akbarzadeh, M.; Mousavi, S.-H.; Tayarani-Najaran, Z.; Atapour-Mashhad, H.; Nikpour, M. Synthesis of new derivatives of 3-aryl-1,5-dimethyl-1H-[1,2,4]triazolo[4′,3′:1,2]pyrimido[4,5-e][1,3,4]oxadiazines as potential antiproliferative agents. J. Heterocycl. Chem. 2011, 48, 183–187. [Google Scholar] [CrossRef]
- Elliott, A.J.; Gibson, M.S. A new synthesis of 4H-1,3,4-benzoxadiazines. J. Chem. Soc. Perkin Trans. 1. 1972, 2915–2917. [Google Scholar] [CrossRef]
- Elliott, A.J.; Gibson, M.S.; Kayser, M.M.; Pawelchak, G.A. Smiles Rearrangement in Hydrazonyl Systems: Extension of a Recent 4H-1,3,4-Benzoxadiazine Synthesis. Can. J. Chem. 1973, 51, 4115–4121. [Google Scholar] [CrossRef]
- Berezin, A.A.; Zissimou, G.; Constantinides, C.P.; Beldjoudi, Y.; Rawson, J.M.; Koutentis, P.A. Route to Benzo- and Pyrido-fused 1,2,4-Triazinyl Radicals via N′-(Het)aryl-N′-[2-nitro(het)aryl]hydrazides. J. Org. Chem. 2014, 79, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Savva, A.C.; Zissimou, G.A.; Mirallai, S.I.; Berezin, A.A.; Demetriades, M.; Constantinides, C.P.; Nicolaides, C.; Trypiniotis, T.; Koutentis, P.A. Preparation of Blatter Radicals via Aza-Wittig Chemistry: The Reaction of N-Arylimino-phosphoranes and 1-(Het)aroyl-2-aryldiazenes. J. Org. Chem. 2017, 82, 7564–7575. [Google Scholar] [CrossRef] [PubMed]
- Keane, L.-A.J.; Mirallai, S.I.; Sweeney, M.; Carty, M.P.; Zissimou, G.A.; Berezin, A.A.; Koutentis, P.A.; Aldabbagh, F. Anti-cancer Activity of Phenyl and Pyrid-2-yl 1,3-Substituted Benzo[1,2,4]triazin-7-ones and Stable Free Radical Precursors. Molecules 2018, 23, 574. [Google Scholar] [CrossRef] [PubMed]
- Karekla, G.; Papagiorgis, P.; Panayi, N.; Zissimou, G.A.; Constantinides, C.P.; Koutentis, P.A.; Itskos, G.; Hayes, S.C. Emission from the stable Blatter radical. New J. Chem. 2017, 41, 8604–8613. [Google Scholar] [CrossRef]
- Constantinides, C.P.; Berezin, A.; Zissimou, G.A.; Manoli, M.; Leitus, G.; Koutentis, P.A. The suppression of columnar π-stacking in 3-adamantyl-1-phenyl-1,4-dihydrobenzo[e][1,2,4]triazin-4-yl. Molecules 2016, 21, 636. [Google Scholar] [CrossRef] [PubMed]
- Morgan, I.S.; Mansikkamäki, A.; Zissimou, G.A.; Koutentis, P.A.; Rouzières, M.; Clérac, R.; Tuononen, H.M. Coordination Complexes of a Neutral 1,2,4-Benzotriazinyl Radical Ligand: Synthesis, Molecular and Electronic Structures, and Magnetic Properties. Chem. Eur. J. 2015, 21, 15843–15853. [Google Scholar] [CrossRef] [PubMed]
- Constantinides, C.P.; Berezin, A.A.; Zissimou, G.A.; Manoli, M.; Leitus, G.M.; Bendikov, M.; Probert, M.R.; Rawson, J.M.; Koutentis, P.A. A Magnetostructural Investigation of an Abrupt Spin-Transition for 1-Phenyl-3-trifluoromethyl-1,4-dihydro-benzo[e][1,2,4]triazin-4-yl. J. Am. Chem. Soc. 2014, 136, 11906–11909. [Google Scholar] [CrossRef] [PubMed]
- Constantinides, C.P.; Berezin, A.A.; Manoli, M.; Leitus, G.M.; Zissimou, G.A.; Bendikov, M.; Rawson, J.M.; Koutentis, P.A. Structural, Magnetic and Computational Correlations of Some Imidazolo-fused 1,2,4-Benzotriazinyl Radicals. Chem. Eur. J. 2014, 20, 5388–5396. [Google Scholar] [CrossRef] [PubMed]
- Demetriou, M.; Berezin, A.A.; Koutentis, P.A.; Krasia-Christoforou, T. Benzotriazinyl-Mediated Controlled Radical Polymerisation of Styrene. Polym. Int. 2014, 63, 674–679. [Google Scholar] [CrossRef]
- Constantinides, C.P.; Berezin, A.A.; Manoli, M.; Leitus, G.M.; Bendikov, M.; Rawson, J.M.; Koutentis, P.A. Effective Exchange Coupling in Alternating-Chains of a π-Extended 1,2,4-Benzotriazin-4-yl. New J. Chem. 2014, 38, 949–954. [Google Scholar] [CrossRef]
- Berezin, A.A.; Koutentis, P.A. Ring contraction of 1,3-diphenylbenzo[1,2,4]triazinyl radicals to 1,2-diphenylbenzimidazoles. Org. Biomol. Chem. 2014, 12, 1641–1648. [Google Scholar] [CrossRef] [PubMed]
- Harwood, L.M. “Dry-Column” Flash Chromatography. Aldrichimica Acta 1985, 18, 25–25. [Google Scholar]
- Herkes, F.E. Synthesis of tetrafluorobenzothiazoles and tetrafluoro-4H-1,3,4-benzothiadiazines. J. Fluorine Chem. 1979, 13, 1–21. [Google Scholar] [CrossRef]
- CrysAlis CCD and CrysAlis RED, version 1.171.32.15; Oxford Diffraction Ltd.: Abingdon/Oxford, UK, 2008.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX suite for single crystal small molecule crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Brandenburg, K. DIAMOND, version 3.1d; Crystal Impact GbR: Bonn, Germany, 2006. [Google Scholar]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zissimou, G.A.; Kourtellaris, A.; Koutentis, P.A. 5,6,7,8-Tetrafluoro-1-(2-nitrophenyl)-3-phenyl-1H-benzo[e][1,3,4]oxadiazine. Molbank 2018, 2018, M997. https://doi.org/10.3390/M997
Zissimou GA, Kourtellaris A, Koutentis PA. 5,6,7,8-Tetrafluoro-1-(2-nitrophenyl)-3-phenyl-1H-benzo[e][1,3,4]oxadiazine. Molbank. 2018; 2018(2):M997. https://doi.org/10.3390/M997
Chicago/Turabian StyleZissimou, Georgia A., Andreas Kourtellaris, and Panayiotis A. Koutentis. 2018. "5,6,7,8-Tetrafluoro-1-(2-nitrophenyl)-3-phenyl-1H-benzo[e][1,3,4]oxadiazine" Molbank 2018, no. 2: M997. https://doi.org/10.3390/M997
APA StyleZissimou, G. A., Kourtellaris, A., & Koutentis, P. A. (2018). 5,6,7,8-Tetrafluoro-1-(2-nitrophenyl)-3-phenyl-1H-benzo[e][1,3,4]oxadiazine. Molbank, 2018(2), M997. https://doi.org/10.3390/M997