4. Materials and Methods
4.1. General Information
All reagents were purchased from Sigma Aldrich, Alfa Aesar, or TCI and used as received without further purification. Solvents were used in RPE grade without further purification. Anhydrous solvents were obtained from a PURESOLV SPS400 apparatus developed by Innovative Technology Inc. (Hong Kong, China). 1H-, 13C-, and 19F-NMR spectra were recorded on a Bruker Avance III 400 MHz instrument (Wissembourg, France). Samples were dissolved in an appropriate deuterated solvent (CDCl3). The chemical shifts (δ) are expressed in ppm relative to internal tetramethylsilane for 1H and 13C nuclei, and coupling constants are indicated in Hz. Abbreviations for signal coupling are as follows: s = singlet; d = doublet; ddt = doublet of doublets of triplets; t = triplet; q = quartet; m = multiplet; br = broad signal. To assign the signals to the different proton and carbon atoms, as well as the relative stereochemistry of the cycloadducts, additional 2D NMR experiments (COSY, HSQC, HMBC) and NOESY experiments were performed. High-resolution mass spectra (HRMS) were performed on Xevo G2-XS Q-TOF WATERS (Milford, MA, USA) by electrospray ionization (ESI) or atmospheric solids analysis probe (ASAP). Infrared (IR) spectra were recorded with a Perkin Elmer 16 PC FTIR ATR spectrometer (Waltham, MA, USA), using the pure product (oil or solid). Thin layer chromatography (TLC) was run on pre-coated aluminum plates of silica gel 60 F-254 (Merck).
4.2. Synthesis of 1
To a solution of 4-hydroxybenzaldehyde (10.0 g, 81.9 mmol, 1.0 eq.) and K
2CO
3 (28.3 g, 204.7 mmol, 2.5 eq.) in CH
3CN (300 mL), 4-bromo-1-butene (12.37 mL, 122.8 mmol, 1.5 eq.) was added. The mixture was refluxed at 80 °C for 48 h (the reaction advancement was monitored by TLC or NMR). After cooling to room temperature, the solvent was removed under reduced pressure. The residue was partitioned between CH
2Cl
2 and water and the aqueous layer was extracted with CH
2Cl
2 (2 × 10 mL). The combined organic extracts were washed with water, dried, and concentrated to afford the product
1 [
15] as a pale yellow oil (13.77 g, 95%).
1H-NMR (400 MHz, CDCl3) δ 9.88 (s, 1H), 7.83 (d, J = 8.7 Hz, 2H), 7.00 (d, J = 8.7 Hz, 2H), 5.90 (ddt, J = 17.1, 10.3, 6.7 Hz, 1H), 5.12–5.21 (m, 2H), 4.10 (t, J = 6.7 Hz, 2H), 2.58 (q, J = 6.7 Hz, 2H). 13C-NMR (100 MHz, CDCl3) δ 191.0, 164.1, 134.0, 132.1, 130.1, 117.6, 114.9, 67.7, 33.5. IR (neat) cm−1 3078, 2938, 2739, 1686, 1598, 1576, 1508, 1250, 1157, 1024. HRMS m/z (ESI): calcd. for C11H13O2 [M + H]+: 177.0916, found: 177.0917.
4.3. Synthesis of
At 0 °C, compound
1 (5.0 g, 28.37 mmol, 1.0 eq.) in a THF/EtOH mixture (1:1 volume ratio, 40 mL) was added dropwise to a stirred solution of NaBH
4 (1.61 g, 42.56 mmol, 1.5 eq.) in a THF/EtOH mixture (1:1 volume ratio, 40 mL). The mixture was stirred at room temperature for 12 h then cooled to 0 °C and quenched with aqueous NH
4Cl solution. The volatiles were evaporated and the residue was extracted with EtOAc (2 × 20 mL). The combined organic extracts were washed with water (2 × 10 mL), dried over MgSO
4, and concentrated under reduced pressure. The product 2 [
15] was obtained as a pale yellow oil (4.62 g, 91%).
1H-NMR (400 MHz, CDCl3) δ 7.28 (d, J = 8.7 Hz, 2H,), 6.89 (d, J = 8.7 Hz, 2H), 5.91 (ddt, J = 17.1, 10.3, 6.7 Hz, 1H), 5.10–5.19 (m, 2H), 4.61 (s, 2H), 4.02 (t, J = 6.7 Hz, 2H), 2.55 (q, J = 6.7 Hz, 2H). 13C-NMR (100 MHz, CDCl3) δ 158.7, 134.5, 133.3, 128.8, 117.2, 114.8, 67.4, 65.2, 33.8. IR (neat) cm−1 3078, 2926, 2872, 1612, 1511, 1241, 1172, 1033, 994, 917. HRMS m/z (ASAP): calcd. for C11H14O2 [M]+: 178.0994, found: 178.0993.
4.4. Synthesis of 3
To a stirred solution of compound 2 (4.0 g, 22.44 mmol, 1.0 eq.) in Et
2O (40 mL) at 0 °C was added slowly a solution of PBr
3 (2.56 mL, 26.93 mmol, 1.2 eq.) in Et
2O (20 mL). The mixture was stirred for 6 h at 0 °C and was stirred at room temperature for another 6 h. The reaction mixture was placed in an acetone ice bath, water was added, and the solution was extracted with diethyl ether. The organic extracts were washed with aqueous NaHCO
3 solution, water then dried over MgSO
4. The solvent was removed under reduced pressure. The product 3 [
15] was obtained as a pale yellow liquid (4.94 g, 91%).
1H-NMR (400 MHz, CDCl3) δ 7.31 (d, J = 8.7 Hz, 2H), 6.88 (d, J = 8.7 Hz, 2H), 5.90 (ddt, J = 17.1, 10.3, 6.7 Hz, 1H), 5.10–5.20 (m, 2H), 4.50 (s, 2H), 4.01 (t, J = 6.7 Hz, 2H), 2.54 (q, J = 6.7 Hz, 2H). 13C-NMR (100 MHz, CDCl3) δ 159.1, 134.4, 130.6, 130.1, 117.3, 115.0, 67.4, 34.2, 33.7. IR (neat) cm−1 3077, 2924, 2870, 1610, 1509, 1246, 1174, 1033, 988, 916. HRMS m/z (ASAP): calcd. for C11H14OBr [M + H]+: 241.0228, found: 241.0229.
4.5. Synthesis of 4
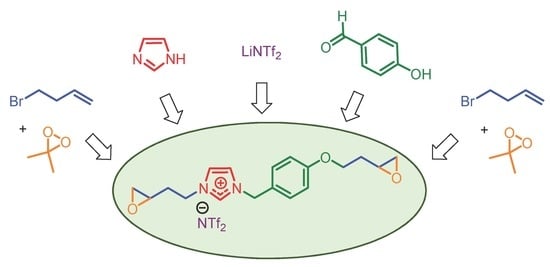
To a solution of imidazole (564 mg, 8.29 mmol, 1.0 eq.) in THF (36 mL), NaH (60%) (664 mg, 16.59 mmol, 2.0 eq.) was added in small portions. The reaction mixture was stirred for 30 min. A solution of compound 3 (2.0 g, 8.29 mmol, 1.0 eq.) in THF (40 mL) was added and the reaction mixture was stirred at 60 °C for 12 h. The reaction was quenched with water (91 mL) and the solvent was removed under reduced pressure. Dichloromethane (92 mL) was added and the organic layer was thoroughly washed two times with water and then separated. The organic layer was dried over MgSO4 and the solvent was removed under vacuum to afford the product 4 as a yellow oil (1.79 g, 95%).
1H-NMR (400 MHz, CDCl3) δ 7.52 (s, 1H), 7.00–7.16 (m, 3H), 6.77–6.97 (m, 3H), 5.89 (ddt, J = 17.1, 10.3, 6.7 Hz, 1H), 4.98–5.22 (m, 4H), 4.00 (t, J = 6.7 Hz, 2H), 2.54 (q, J = 6.7 Hz, 2H). 13C-NMR (100 MHz, CDCl3) δ 159.0, 137.4, 134.4, 129.9, 129.0, 128.3, 119.2, 117.3, 115.1, 67.4, 50.5, 33.7. IR (neat) cm−1 3077, 2928, 2872, 1612, 1512, 1243, 1176, 1074, 1029, 905. HRMS m/z (ESI): calcd. for C14H17N2O [M + H]+: 229.1341, found: 229.1343.
4.6. Synthesis of 5
A solution of compound 4 (1.75 g, 7.65 mmol, 1.0 eq.) and 4-bromo-1-butene (0.77 mL, 7.65 mmol, 1.0 eq.) in CH3CN (50 mL) was stirred at 80 °C. After 48 h, the reaction mixture was cooled to room temperature and the solvent was removed under reduced pressure. The product 5 was obtained as a yellow oil (2.60 g, 94%).
1H-NMR (400 MHz, CDCl3) δ 10.65 (s, 1H), 7.39 (d, J = 8.7 Hz, 2H), 7.30–7.34 (m, 1H), 7.21–7.25 (m, 1H), 6.88 (d, J = 8.7 Hz, 2H), 5.72–5.94 (m, 2H), 5.48 (s, 2H), 4.99–5.22 (m, 4H), 4.42 (t, J = 6.7 Hz, 2H), 3.98 (t, J = 6.7 Hz, 2H), 2.66 (q, J = 6.7 Hz, 2H), 2.52 (q, J = 6.7 Hz, 2H). 13C-NMR (100 MHz, CDCl3) δ 159.9, 137.3, 134.2, 132.3, 130.7, 124.9, 122.1, 121.5, 119.9, 117.4, 115.5, 67.4, 53.1, 49.4, 34.5, 33.6. IR (neat) cm−1 3075, 2980, 1612, 1559, 1513, 1247, 1179, 1154, 1031, 919. HRMS m/z (ESI): calcd. for C18H23N2O [M]+: 283.1810, found: 283.1811.
4.7. Synthesis of 6
To a solution of compound 5 (2.52 g, 6.94 mmol, 1.0 eq.) in H2O (150 mL), bis(trifluoromethane)sulfonimide lithium salt (2.19 g, 7.64 mmol, 1.1 eq.) was added. The solution was stirred at room temperature for 24 h and then extracted with dichloromethane. The organic layer was washed several times with water, dried over MgSO4, and concentrated under reduced pressure. The product 6 was obtained as a yellow oil (3.46 g, 89%).
1H-NMR (400 MHz, CDCl3) δ 8.83 (s, 1H), 7.20–7.30 (m, 3H), 7.13–7.17 (m, 1H), 6.92 (d, J = 8.7 Hz, 2H), 5.89 (ddt, J = 17.1, 10.3, 6.7 Hz, 1H), 5.73 (ddt, J = 17.0, 10.2, 6.7 Hz, 1H), 4.98–5.28 (m, 6H), 4.26 (t, J = 6.7 Hz, 2H), 4.01 (t, J = 6.7 Hz, 2H), 2.50–2.63 (m, 4H). 13C-NMR (100 MHz, CDCl3) δ 160.2, 135.7, 134.3, 132.1, 130.6, 124.2, 122.4, 121.9, 120.1, 120.0 (q, JCF = 322.4 Hz), 117.4, 115.6, 67.5, 53.4, 49.6, 34.4, 33.6. 19F-NMR (376 MHz, CDCl3) δ −78.9. IR (neat) cm−1 3148, 3086, 2925, 1614, 1562, 1515, 1348, 1178, 1133, 1053. HRMS m/z (ESI): calcd. for C18H23N2O [M]+: 283.1810, found: 283.1811.
4.8. Synthesis of 7
To a solution of compound
6 (50 mg, 0.0887 mmol, 1.0 eq.) in acetone (1.00 mL), freshly prepared dimethyldioxirane (0.045 mol/L) (5.52 mL, 0.2484 mmol, 2.8 eq.) was added [
15]. The reaction mixture was stirred at room temperature for 2 h (
1H-NMR monitoring). Two drops of DMS was added to quench the reaction mixture and neutralized the excess of dimethyldioxirane. The crude was concentrated under reduced pressure. The product
7 was obtained as a yellow oil (53 mg, 100%).
1H-NMR (400 MHz, CDCl3) δ 8.98 (s, 1H), 7.28–7.33 (m, 3H), 7.11–7.13 (m, 1H), 6.95 (d, J = 8.7 Hz, 2H), 5.28 (s, 2H), 4.41 (t, J = 6.7 Hz, 2H), 4.09–4.17 (m, 2H), 3.12–3.18 (m, 1H), 2.98–3.04 (m, 1H), 2.78–2.86 (m, 2H), 2.42–2.60 (m, 3H), 2.10–2.20 (m, 1H), 1.75–1.96 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ 160.1, 136.2, 130.7, 124.1, 122.8, 121.7, 120.0 (q, JCF = 322.4 Hz), 115.7, 65.0, 53.6, 49.7, 49.3, 47.9, 47.3, 46.7, 32.7, 32.5. 19F-NMR (376 MHz, CDCl3) δ −78.8. IR (neat) cm−1 3149, 2934, 1613, 1563, 1516, 1348, 1330, 1178, 1132, 1052. HRMS m/z (ESI): calcd. for C18H23N2O3 [M]+: 315.1709, found: 315.1712.



{kind=link}
{kind=link}
