3.1. General
Analytical data for previously reported compounds matched those already described and are cited in the preceding text. 1H-NMR spectra were recorded on a Bruker Avance 300 (Coventry, UK), (300 MHz) instrument or Bruker Avance 400 (400 MHz) instrument using deuterated chloroform (or other indicated solvent) as reference. The chemical shift data for each signal are given as δ in units of parts per million (ppm) relative to tetramethylsilane (TMS) where δ = 0.00 ppm. The multiplicity of each signal is indicated by: s (singlet), bs (broad singlet), d (doublet), dd (doublet of doublets), t (triplet), appt (apparent triplet), m (multiplet). The number of protons (n) for a given resonance is indicated by nH. Coupling constants (J) are quoted in Hz and recorded to the nearest 0.1 Hz. 13C-NMR spectra were recorded on a Bruker Avance 300 (75 MHz) instrument or Bruker Avance 400 (100 MHz) instrument. The chemical shift data for each signal are given as δ in units of parts per million (ppm) relative to tetramethylsilane (TMS) where δ = 0.00 ppm. FT-IR spectra were recorded on a Thermo Scientific Nicolet iS10 instrument (Stafford, UK). Analytical thin layer chromatography (TLC) was carried out on pre-coated 0.25 mm Merck (Nottingham, UK) KGaA 60 F254 silica gel plates. Visualisation was by adsorption of UV light, or thermal development after dipping in a methanolic solution of sulfuric acid (5% v/v), vanillin or KMnO4. Chemicals were purchased from Acros/Fisher (Loughborough, UK), Aldrich (Gillingham, UK) or Alfa Aesar (Heysham, UK). All solvents and reagents were purified and dried where necessary. Where appropriate and if not stated otherwise, all non-aqueous reactions were performed under an inert atmosphere of nitrogen, using a vacuum manifold with nitrogen passed through molecular sieves. Brine refers to a saturated aqueous solution of sodium chloride. Hexane refers to the fraction from petroleum ether boiling between 40 and 60 °C. RT refers to room temperature.
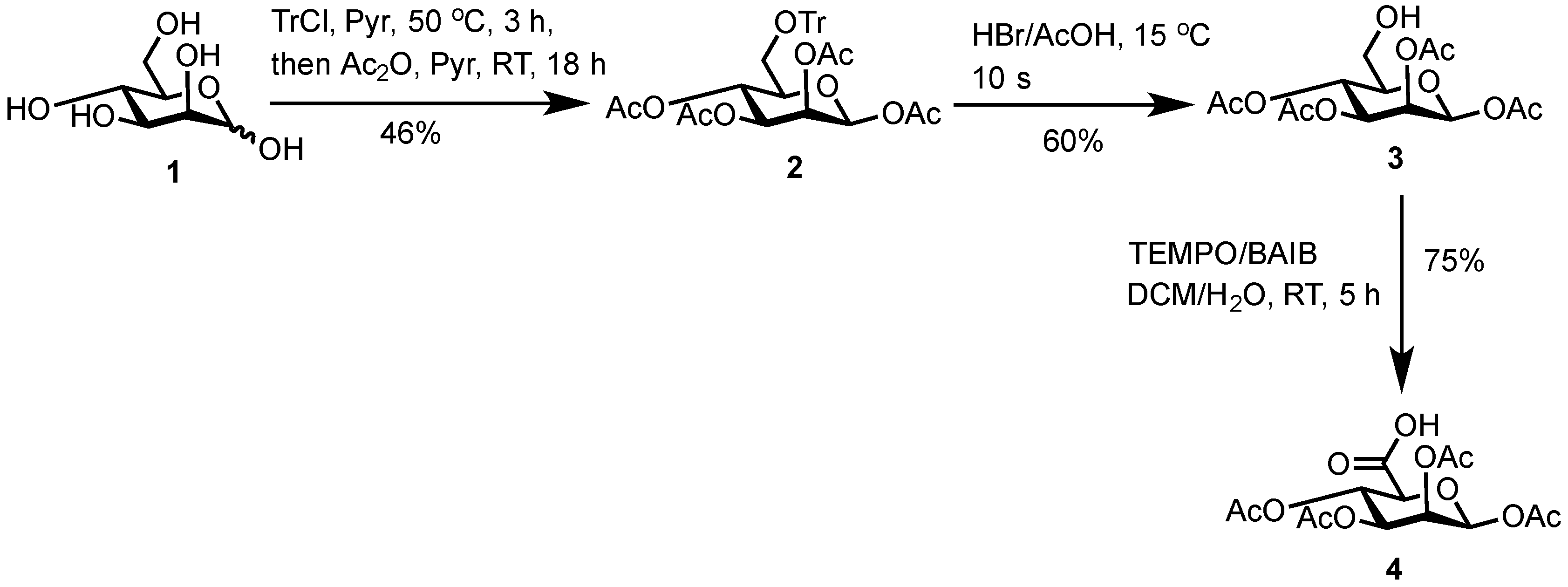
3.2. 1,2,3,4-Tetra-O-Acetyl-6-O-Triphenylmethyl-β-d-Mannopyranose (2)
d-Mannose (
1) (25.8 g, 142 mmol, 1.00 equiv.) and trityl chloride (40.1 g, 143.0 mmol, 1.03 equiv.) were stirred in anhydrous pyridine (125 mL) under a N
2 atmosphere. The reaction mixture was heated to 50 °C and stirred for 3 h. The solution was cooled to RT, placed in an ice bath and acetic anhydride (75 mL) added via a dropping funnel over 30 min. The solution was then stirred at RT for 18 h, whereupon TLC analysis (hexane/ethyl acetate, 3/1) showed complete conversion of starting material to a higher R
f value spot. The reaction mixture was poured onto ice (750 mL) and stirred vigorously for 1.5 h to afford a white solid (water was exchanged twice during this time to remove pyridine). The solid was collected by filtration, washed with ice-cold water (250 mL) and dried for 2.5 h under vacuum. The solid was recrystallized from the minimum amount of hot ethanol, collected by filtration and dried over P
2O
5 in a vacuum desiccator to afford 1,2,3,4-tetra-
O-acetyl-6-
O-triphenylmethyl-β-
d-mannopyranose (
2) as a white solid (39.0 g, 66.0 mmol, 46%).
1H-NMR,
13C-NMR and MS collected for (
2) were in good agreement with those already reported [
8].
3.3. 1,2,3,4-Tetra-O-Acetyl-β-d-Mannopyranose (3)
1,2,3,4-tetra-
O-acetyl-6-
O-triphenylmethyl-β-
d-mannopyranose (
2) (10.2 g, 17.3 mmol, 1.00 equiv.) was dissolved in acetic acid (120 mL) at 40 °C with stirring. The solution was cooled to 15 °C and HBr/acetic acid (33%
w/
w in acetic acid, 5.0 mL, 20.4 mmol, 1.20 equiv.) was added dropwise, forming a yellow precipitate almost immediately. After 10 s, the solution was filtered under vacuum through Celite
TM onto water (250 mL) and the solids washed with acetic acid (10 mL). The flitrate was quenched slowly with saturated aqueous NaHCO
3 solution (250 mL), forming an opaque white suspension which was extracted with chloroform (2 × 200 mL). The organic layers were combined, washed with water (100 mL), saturated aqueous NaHCO
3 solution (100 mL), dried (MgSO
4), filtered and concentrated in vacuo to afford the crude product as an oil. TLC analysis (hexane/ethyl acetate, 3/1) showed complete conversion of starting material to a lower R
f value spot and a higher UV active R
f value spot corresponding to trityl bromide. The crude product was dissolved in the minimum amount of hot chloroform and induced to crystallise by addition of diethyl ether. The crystals formed were collected by filtration and dried in vacuo, affording 1,2,3,4-tetra-
O-acetyl-β-
d-mannopyranose (
3) as a white solid (3.6 g, 10.4 mmol, 60%).
1H-NMR,
13C-NMR and MS collected for (
3) were in good agreement with those already reported [
8].
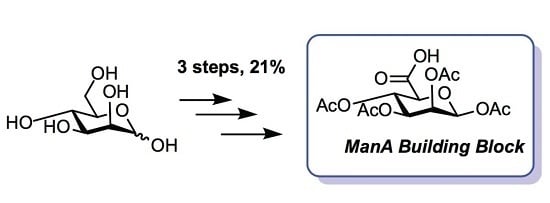
3.4. 1,2,3,4-Tetra-O-Acetyl-β-d-Mannuronic Acid (4)
To a vigorously stirred solution of 1,2,3,4-tetra-O-acetyl-β-d-mannopyranose (3) (1.5 g, 4.30 mmol, 1.0 equiv.) in dichloromethane (20 mL) and water (10 mL) was added (2,2,6,6-tetramethylpiperidin-1-yl)oxyl radical (TEMPO) (336 mg, 2.2 mmol, 0.50 equiv.) and [bis(acetoxy)iodo]benzene (BAIB) (6.9 g, 21.5 mmol, 5.00 equiv.). The solution was stirred at RT for 5 h, whereupon TLC analysis (hexane/ethyl acetate, 1/1) showed complete conversion of starting material to a baseline Rf value spot. The reaction was quenched with aqueous Na2S2O3 solution (10% w/v, 30 mL), the organic layer removed and the aqueous layer acidified to pH 3 using 1M HCl, followed by extraction with ethyl acetate (2 × 30 mL). The organic extracts were combined, dried (MgSO4), filtered and concentrated in vacuo to afford 1,2,3,4-tetra-O-acetyl-β-d-mannuronic acid (4) as a white solid (1.2 g, 3.3 mmol, 75%). Rf = 0.81 (dichloromethane/methanol, 9/1); 1H-NMR (300 MHz, CDCl3) δH 5.96 (1H, d, J = 1.5 Hz, H1), 5.50 (1H, dd, J = 3.2, 1.5 Hz, H2), 5.48 (1H, appt, J = 9.1 Hz, H4), 5.21 (1H, dd, J = 9.1, 3.2 Hz, H3), 4.2 (1H, d, J = 9.1 Hz, H5), 2.21 (3H, s, C(O)CH3), 2.13 (3H, s, C(O)CH3), 2.10 (3H, s, C(O)CH3), 2.04 (3H, s, C(O)CH3). 13C-NMR (100 MHz, CDCl3) δC 169.9 (C=O), 169.7 (C=O), 169.5 (C=O), 169.4 (C=O), 168.2 (C=O), 89.4 (C1), 72.3 (C5), 69.3 (C3), 66.9 (C2), 65.9 (C4), 20.4 (C(O)CH3), 20.4 (C(O)CH3), 20.3 (C(O)CH3), 20.2 (C(O)CH3); FT-HRMS NSI (ES−) m/z Found: (M−H)− 361.0767, C14H17O11 requires 361.0776; FT-IR vmax/cm−1 2986 (O–H), 1751 (C=O).


{kind=link}
{kind=link}
