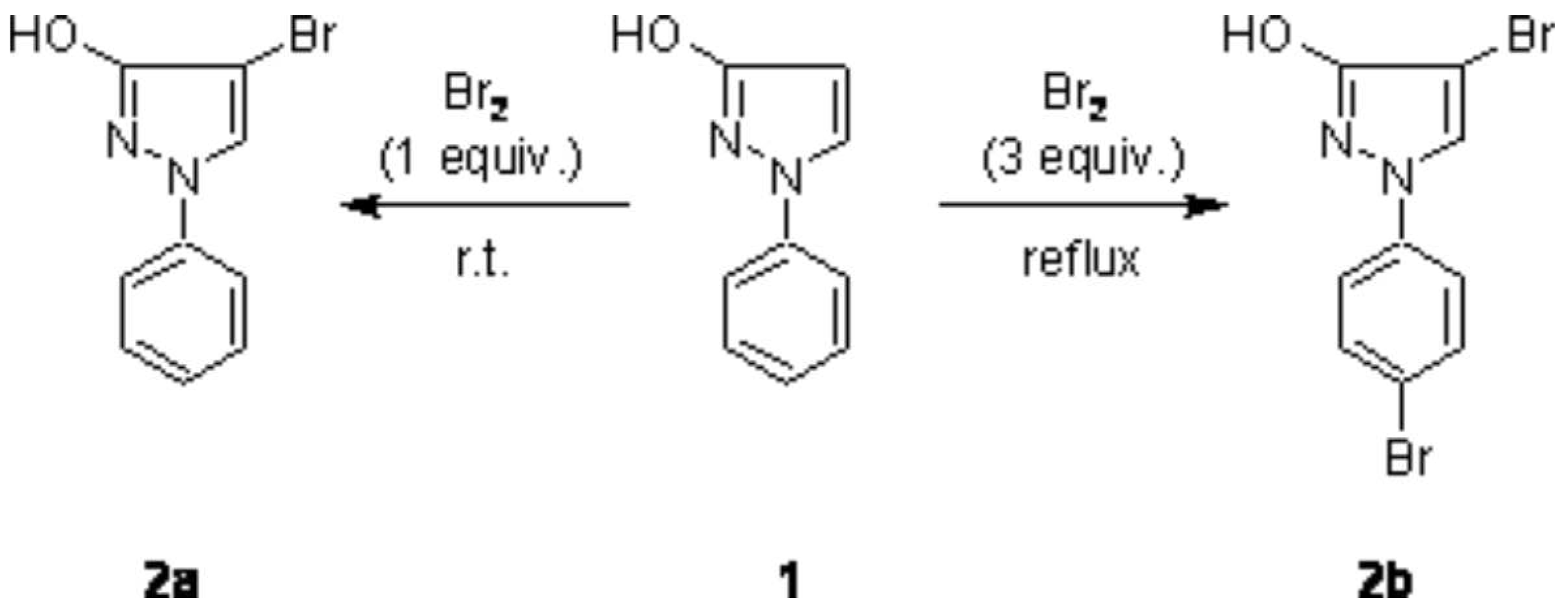
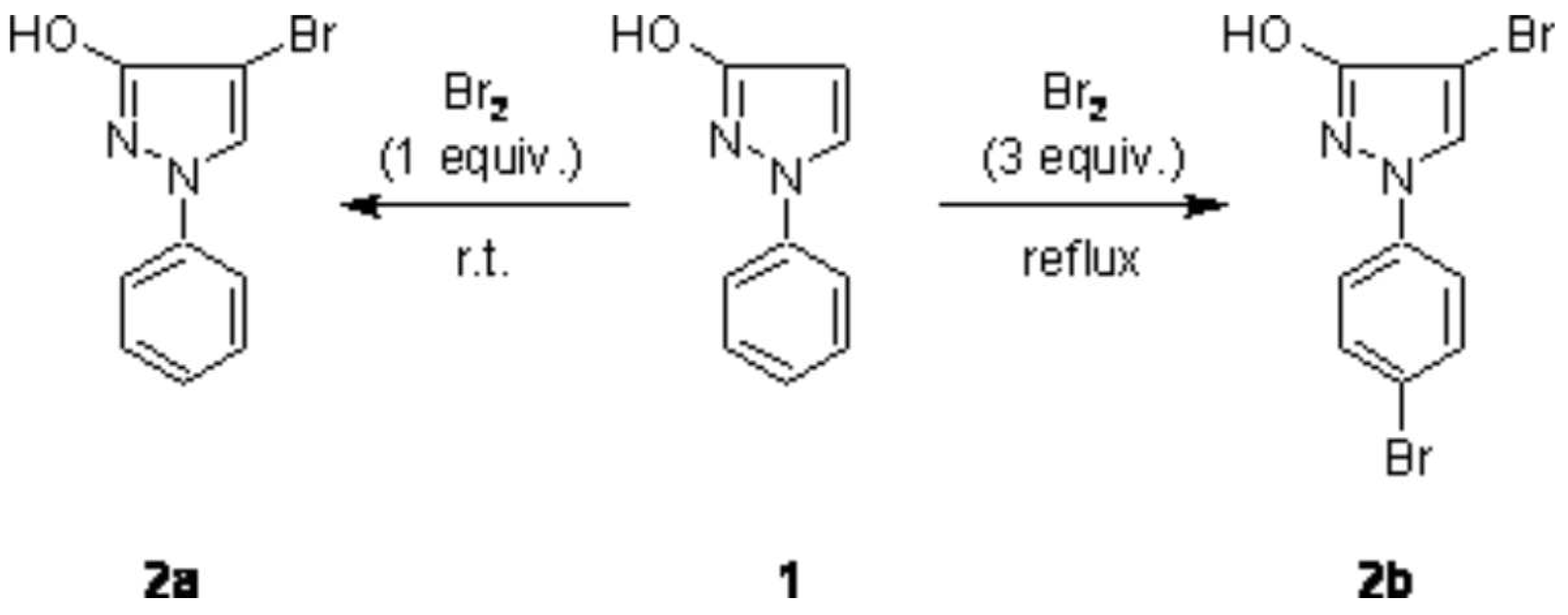
In the course of investigations regarding Pd-catalyzed cross coupling reactions with halopyrazoles [
1], we prepared 4-bromo-1-phenylpyrazol-3-ol (2a) via reaction of 1-phenylpyrazol-3-ol (1) with one equivalent of bromine in high yield similarly to a previously described procedure [
2]. In this respect it seemed interesting for us, if treatment of 1 with an excess of Br
2 would lead to a dibromination product (introduction of a bromo atom also into the phenyl ring) and thus to a synthon with two different targets for further C–C bond formation. Such a behaviour was reported in a related case, namely in the bromination of 1-phenylpyrazole, where the use of excess bromine together with severe reaction conditions was found to give 15% 4-bromo-1-(4-bromphenyl)pyrazole besides the main product 4-bromo-1-phenylpyrazole (65%) [
3].
We found that treatment of 1 with 3 equivalents of bromine in a solution of boiling carbon tetrachloride gave the dibromination product 2b in 82% yield (
Scheme 1). The structure of 2b was unambiguously determined by spectroscopic methods (
1H,
13C, and
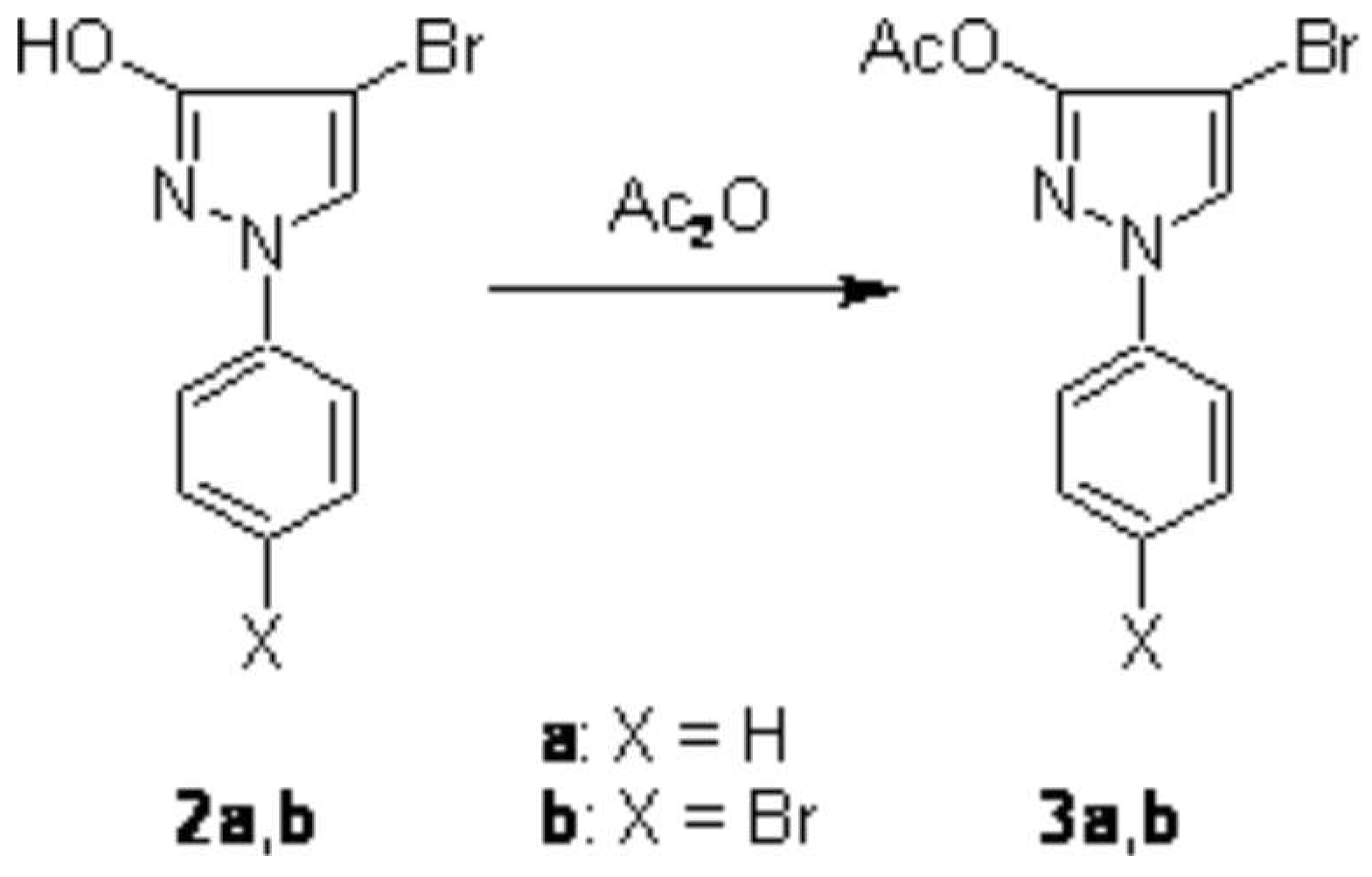
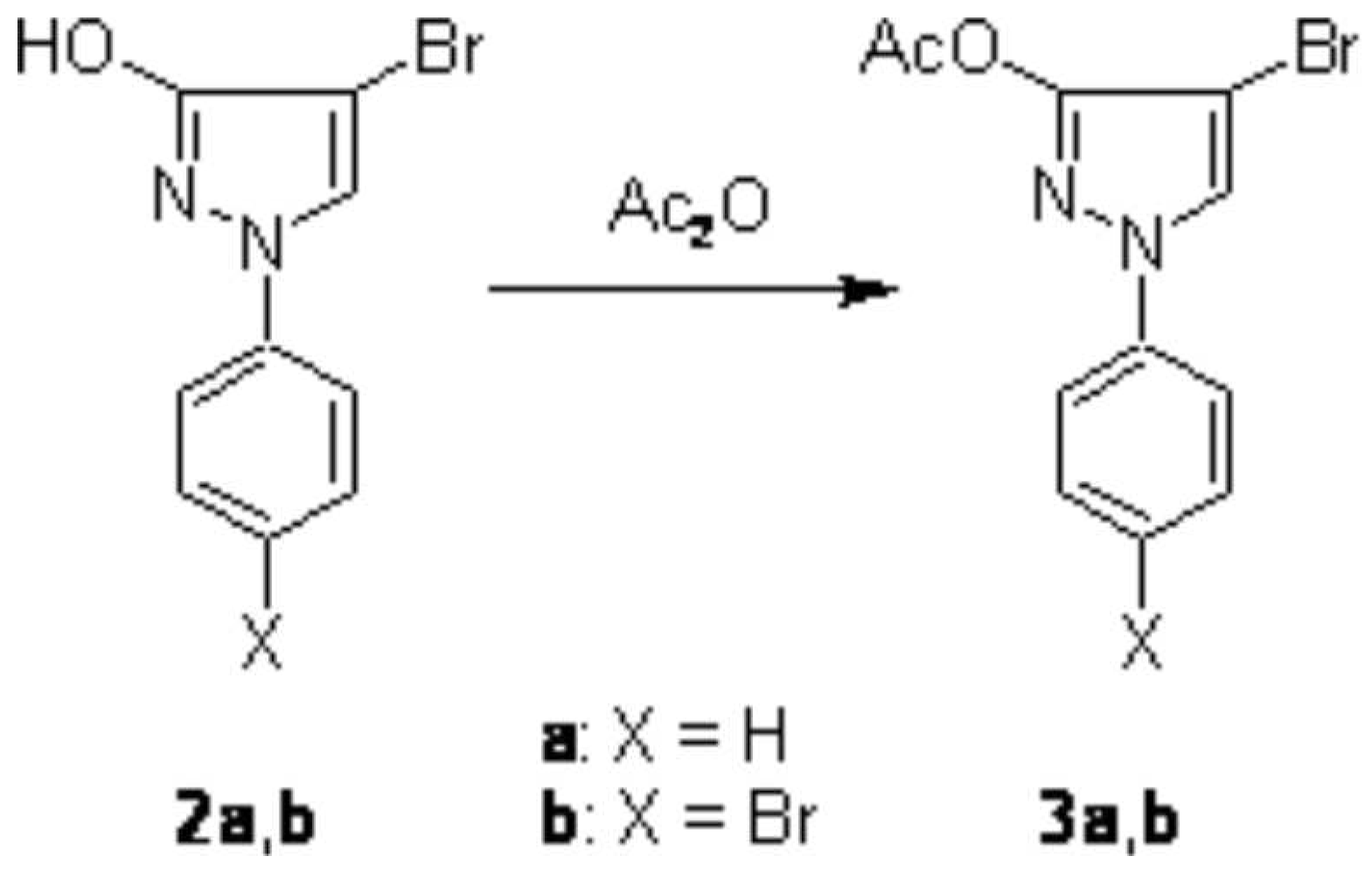
15N NMR, MS) and CHN analysis. Furthermore, treatment of 2a and 2b with boiling acetic anhydride afforded the corresponding
O-acetyl products 3a and 3b in high yield (
Scheme 2).
4-Bromo-1-(4-bromophenyl)-1H-pyrazol-3-ol (2b)
To a well stirred solution of hydroxypyrazole 1 [
4] (1 g, 6.25 mmol) in carbon tetrachloride (30 mL) a solution of bromine (0.32 mL, 6.25 mmol) in carbon tetrachloride (8 mL) was added dropwise during 1 h, and stirring was continued for 2 h at room temperature. Then the reaction temperature was increased to reach reflux temperature within 1 h while adding a second equivalent of bromine (0.32 mL, 6.25 mmol) in carbon tetrachloride (8 mL). After the addition was complete, stirring was continued for further 5 h at reflux temperature. Then another equivalent of bromine (0.32 mL, 6.25 mmol) in carbon tetrachloride (8 mL) was added dropwise during 1 h, and stirring was continued for further 9 h at reflux temperature. The reaction mixture was then allowed to cool to room temperature; the precipitated solid was filtered off, washed with carbon tetrachloride (15 mL), and recrystallized from aqueous ethanol to afford 1.63 g (82%) of pure 2b.
Melting point: 199–201 °C, yellowish crystals.
1H NMR (300 MHz, DMSO-
d6) [
6]: δ (ppm) 11.07 (s, 1H, OH), 8.54 (s, 1H, H-5), 7.62 (m, 4H, Ph H-2,3,5,6).
13C NMR (75 MHz, DMSO-
d6) [
6]: δ (ppm) 159.6 (C-3,
3J(C-3,H-5) = 8.8 Hz), 138.5 (Ph C-1), 132.2 (Ph C-3,5), 128.7 (C-5,
1J = 195.0 Hz), 118.6 (Ph C-2,6), 117.3 (Ph C-4), 82.7 (C-4,
2J(C-4,H-5) = 5.0 Hz).
15N NMR (50 MHz, DMSO-
d6) [
7]: δ (ppm) −119.4 (N-2), −189.4 (N-1).
MS (m/z, %) [
8]: 320 (M
+, 49), 318 (M
+, 100), 316 (M
+, 45), 239 (22), 237 (21), 157 (23), 157 (23), 155 (19).
Elemental Analysis: Calculated for C9H6Br2N2O (317.96): C, 34.00%; H, 1.90%; N, 8.81%. Found: C, 34.24%; H, 1.95%; N, 8.64%.
4-Bromo-1-phenyl-1H-pyrazol-3-yl acetate (3a)
Bromopyrazole 2a (1.91 g, 8 mmol) – which had been prepared by treatment of hydroxypyrazole 1 with one equivalent of bromine at room temperature following a known procedure [
2] – and excess acetic anhydride (25 mL) were heated to reflux for 30 min. Then, H
2O (15 mL) was added and the solution was stirred for further 30 min. The mixture was poured into ice-water (50 mL) and stirred for 30 min. Then the precipitate was filtered off, washed with H
2O, and dried to give 2.07 g (92%) of pure 3a.
Melting point: 82–85 °C, off-white crystals.
IR (KBr) [
5]: 1758 cm
−1 (C=O).
1H NMR (300 MHz, CDCl
3) [
6]: δ (ppm) 7.90 (s, 1H, H-5), 7.57 (m, 2H, Ph H-2,6), 7.43 (m, 2H, Ph H-3,5), 7.29 (m, 1H, Ph H-4), 2.38 (s, 3H, CH
3).
13C NMR (75 MHz, CDCl
3) [
6]: δ (ppm) 167.6 (CO,
2J(CO,CH
3) = 7.1 Hz), 154.0 (C-3,
3J(C-3,H-5) = 9.4 Hz), 139.2 (Ph C-1), 129.5 (Ph C-3,5), 128.4 (C-5,
1J = 194.3 Hz), 127.0 (Ph C-4), 118.5 (Ph C-2,6), 87.7 (C-4,
2J(C-4,H-5) = 5.1 Hz), 20.4 (CH
3,
1J = 131.0 Hz).
15N NMR (50 MHz, CDCl
3) [
7]: δ (ppm) −177.0 (N-1); N-2 not found.
MS (m/z, %) [
8]: 282 (M
+, 6), 280 (M
+, 6), 240 (100), 238 (99), 104 (49), 77 (84), 51 (30), 43 (52).
Elemental Analysis: Calculated for C11H9BrN2O2 (281.11): C, 47.00%; H, 3.23%; N, 9.97%. Found: C, 46.74%; H, 3.07%; N, 9.84%.
4-Bromo-1-(4-bromophenyl)-1H-pyrazol-3-yl acetate (3b)
Dibromopyrazole 2b (1.27 g, 4 mmol) and excess acetic anhydride (15 mL) were refluxed for 30 min. Then H2O (10 mL) was added and the solution was stirred for further 30 min. The mixture was poured into ice-water (50 mL) and stirred for 30 min. Then the precipitate was filtered off, washed with H2O, and dried to give 1.17 g (81%) of pure 3b.
Melting point: 77 °C, beige powder.
IR (KBr) [
5]: 1780, 1761 cm
−1 (C=O).
1H NMR (300 MHz, CDCl
3) [
6]: δ (ppm) 7.87 (s, 1H, H-5), 7.55 (m, 2H, Ph H-3,5), 7.46 (m, 2H, Ph H-2,6), 2.38 (s, 3H, CH
3).
13C NMR (75 MHz, CDCl
3) [
6]: δ (ppm) 167.5 (CO,
2J(CO,CH
3) = 7.1 Hz), 154.4 (C-3,
3J(C-3,H-5) = 9.4 Hz), 138.3 (Ph C-1), 132.6 (Ph C-3,5), 128.3 (C-5,
1J = 194.3 Hz), 120.4 (Ph C-4), 120.0 (Ph C-2,6), 88.4 (C-4,
2J(C-4,H-5) = 5.0 Hz), 20.4 (CH
3,
1J = 130.7 Hz).
15N NMR (50 MHz, CDCl
3) [
7]: δ (ppm) −99.2 (N-2), −179.3 (N-1).
MS (m/z, %) [
8]: 362 (M
+, 1), 360 (M
+, 3), 358 (M
+, 1), 320 (28), 318 (60), 316 (28), 157 (26), 155 (25), 76 (29), 75 (30), 43 (100).
Elemental Analysis: Calculated for C11H8Br2N2O2 (360.00): C, 36.70%; H, 2.24%; N, 7.78%. Found: C, 36.82%; H, 2.19%; N, 7.76%.

{kind=link}
{kind=link}