Targeting Receptor Tyrosine Kinases for Chemoprevention by Green Tea Catechin, EGCG
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
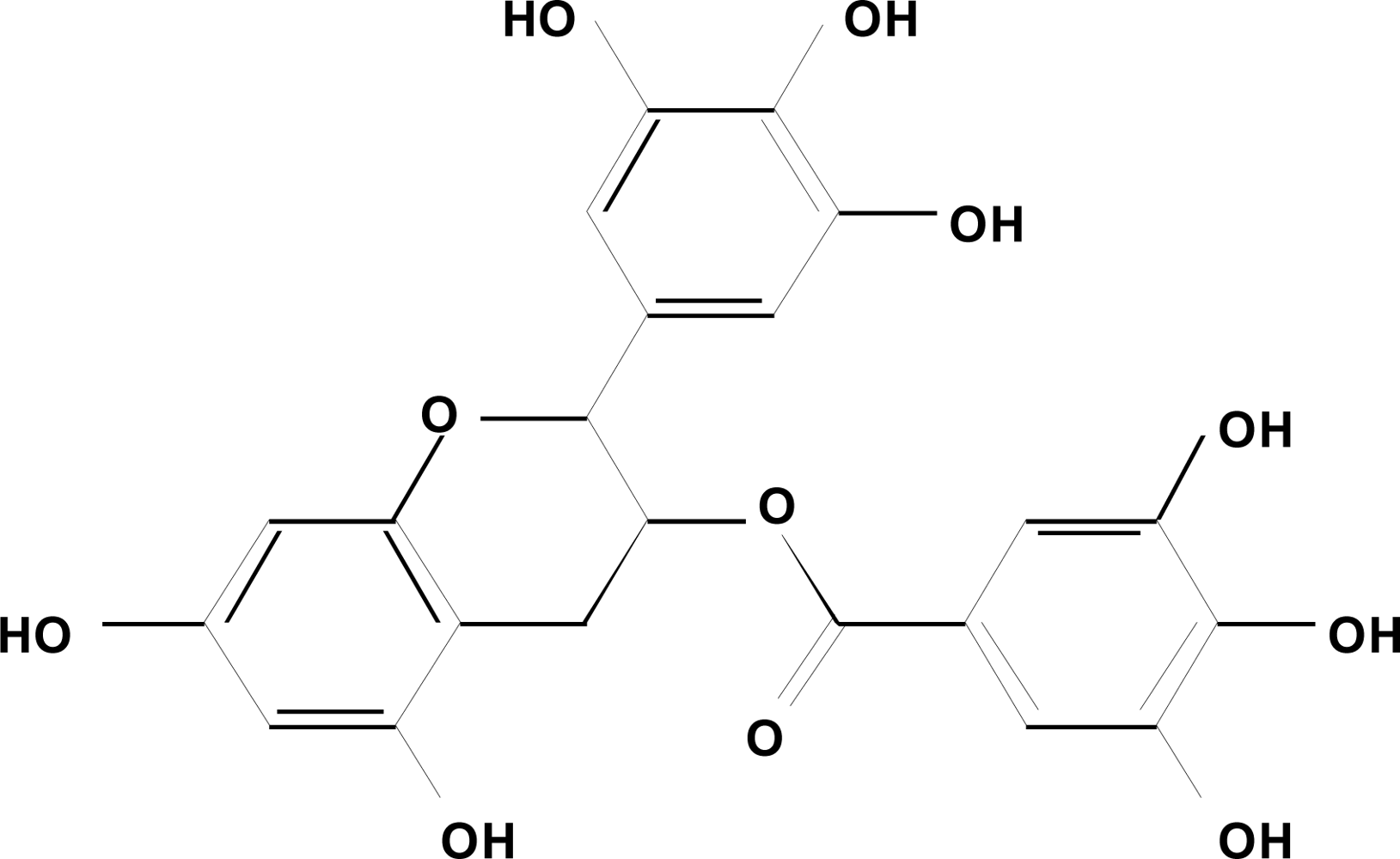
2. Green Tea and Cancer Chemoprevention
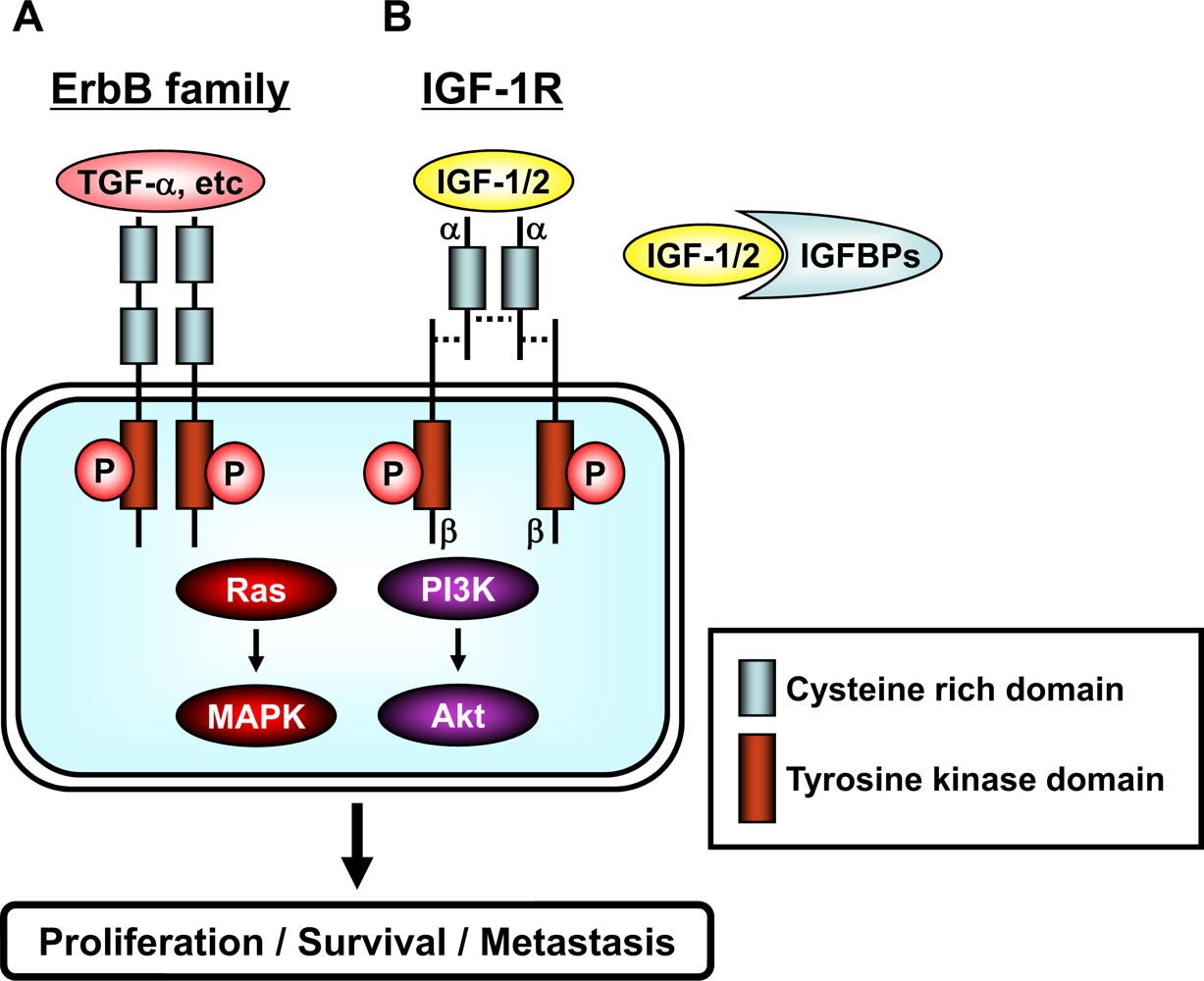
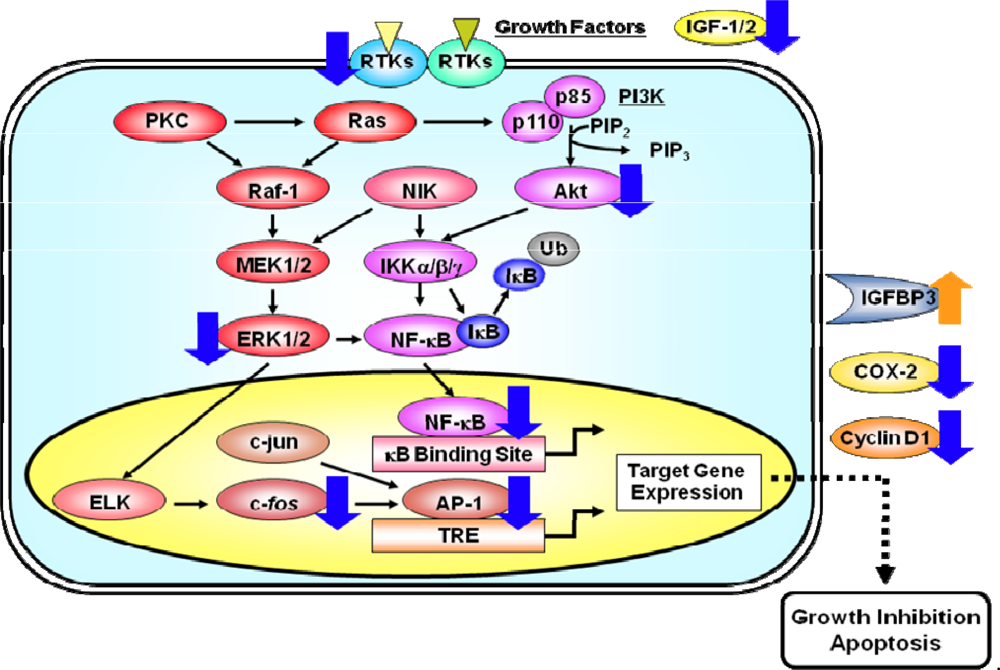
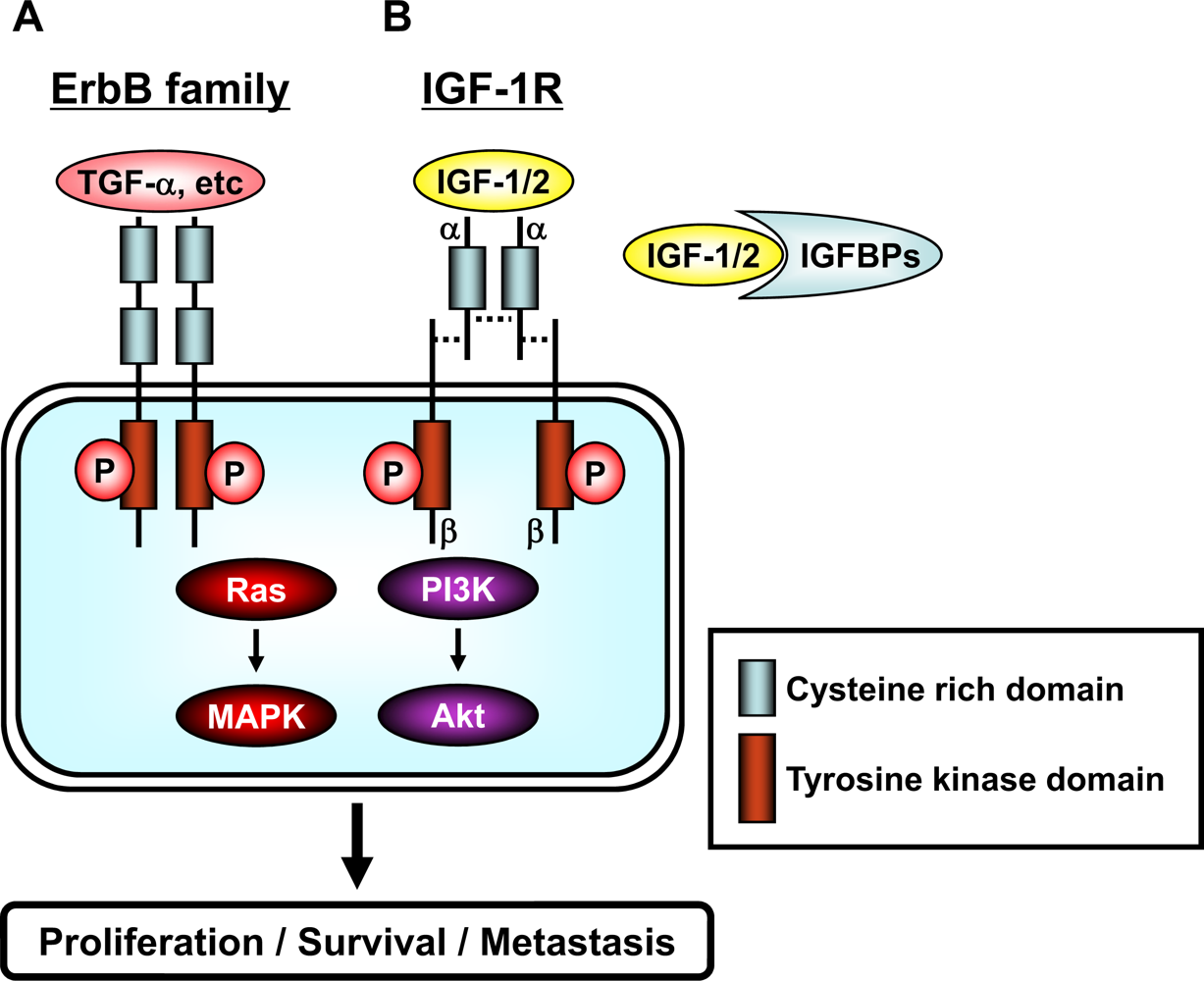
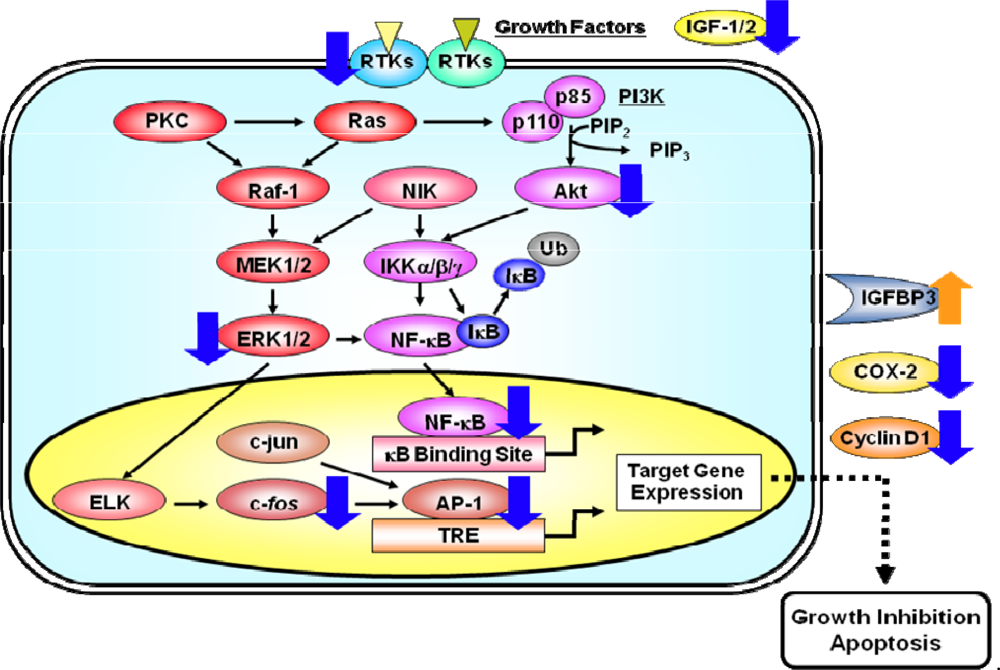
3. Membrane Associated RTKs, their Downstream Signaling Pathways, and Transcription Factors
4. Effects of EGCG on the erbB Family of RTKs
5. Effects of EGCG on the IGF/IGF-1R and VEGF/VEGFR Systems
6. Direct Effects of EGCG on Signaling Pathways and Transcription Factors
7. Effects of EGCG on Expression Levels of Cyclin D1 and COX-2
8. Lipid Rafts: a New Target of EGCG
9. Future Perspectives: Possibility for Clinical Application of EGCG
10. Conclusions
References
- Donaldson, MS. Nutrition and cancer: a review of the evidence for an anti-cancer diet. Nutr. J. 2004, 3, 19. [Google Scholar] [Green Version]
- Surh, YJ. Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer 2003, 3, 768–780. [Google Scholar] [Green Version]
- Hanausek, M; Walaszek, Z; Slaga, TJ. Detoxifying cancer causing agents to prevent cancer. Integr. Cancer Ther. 2003, 2, 139–144. [Google Scholar] [Green Version]
- Hursting, SD; Slaga, TJ; Fischer, SM; DiGiovanni, J; Phang, JM. Mechanism-based cancer prevention approaches: targets, examples, and the use of transgenic mice. J. Natl. Cancer Inst. 1999, 91, 215–225. [Google Scholar] [Green Version]
- Shimizu, M; Weinstein, IB. Modulation of signal transduction by tea catechins and related phytochemicals. Mutat. Res. 2005, 591, 147–160. [Google Scholar] [Green Version]
- Khan, N; Afaq, F; Saleem, M; Ahmad, N; Mukhtar, H. Targeting multiple signaling pathways by green tea polyphenol (–)-epigallocatechin-3-gallate. Cancer Res. 2006, 66, 2500–2505. [Google Scholar] [Green Version]
- Wang, J; Eltoum, IE; Lamartiniere, CA. Genistein alters growth factor signaling in transgenic prostate model (TRAMP). Mol. Cell. Endocrinol. 2004, 219, 171–180. [Google Scholar] [Green Version]
- Korutla, L; Cheung, JY; Mendelsohn, J; Kumar, R. Inhibition of ligand-induced activation of epidermal growth factor receptor tyrosine phosphorylation by curcumin. Carcinogenesis 1995, 16, 1741–1745. [Google Scholar] [Green Version]
- Yang, CS; Maliakal, P; Meng, X. Inhibition of carcinogenesis by tea. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 25–54. [Google Scholar] [Green Version]
- Shimizu, M; Deguchi, A; Lim, JT; Moriwaki, H; Kopelovich, L; Weinstein, IB. (–)-Epigallocatechin gallate and polyphenon E inhibit growth and activation of the epidermal growth factor receptor and human epidermal growth factor receptor-2 signaling pathways in human colon cancer cells. Clin. Cancer Res. 2005, 11, 2735–2746. [Google Scholar] [Green Version]
- Shimizu, M; Deguchi, A; Joe, AK; McKoy, JF; Moriwaki, H; Weinstein, IB. EGCG inhibits activation of HER3 and expression of cyclooxygenase-2 in human colon cancer cells. J. Exp. Ther. Oncol. 2005, 5, 69–78. [Google Scholar] [Green Version]
- Shimizu, M; Deguchi, A; Hara, Y; Moriwaki, H; Weinstein, IB. EGCG inhibits activation of the insulin-like growth factor-1 receptor in human colon cancer cells. Biochem. Biophys. Res. Commun. 2005, 334, 947–953. [Google Scholar] [Green Version]
- Masuda, M; Suzui, M; Weinstein, IB. Effects of epigallocatechin-3-gallate on growth, epidermal growth factor receptor signaling pathways, gene expression, and chemosensitivity in human head and neck squamous cell carcinoma cell lines. Clin. Cancer Res. 2001, 7, 4220–4229. [Google Scholar] [Green Version]
- Masuda, M; Suzui, M; Lim, JT; Deguchi, A; Soh, JW; Weinstein, IB. Epigallocatechin-3-gallate decreases VEGF production in head and neck and breast carcinoma cells by inhibiting EGFR-related pathways of signal transduction. J. Exp. Ther. Oncol. 2002, 2, 350–359. [Google Scholar] [Green Version]
- Masuda, M; Suzui, M; Lim, JT; Weinstein, IB. Epigallocatechin-3-gallate inhibits activation of HER-2/neu and downstream signaling pathways in human head and neck and breast carcinoma cells. Clin. Cancer Res. 2003, 9, 3486–3491. [Google Scholar] [Green Version]
- Shimizu, M; Shirakami, Y; Sakai, H; Tatebe, H; Nakagawa, T; Hara, Y; Weinstein, IB; Moriwaki, H. EGCG inhibits activation of the insulin-like growth factor (IGF)/IGF-1 receptor axis in human hepatocellular carcinoma cells. Cancer Lett. 2008, 262, 10–18. [Google Scholar] [Green Version]
- Adachi, S; Nagao, T; Ingolfsson, HI; Maxfield, FR; Andersen, OS; Kopelovich, L; Weinstein, IB. The inhibitory effect of (–)-epigallocatechin gallate on activation of the epidermal growth factor receptor is associated with altered lipid order in HT29 colon cancer cells. Cancer Res. 2007, 67, 6493–6501. [Google Scholar] [Green Version]
- Herbst, RS; Fukuoka, M; Baselga, J. Gefitinib--a novel targeted approach to treating cancer. Nat. Rev. Cancer 2004, 4, 956–965. [Google Scholar] [Green Version]
- Hynes, NE; Lane, HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [Green Version]
- Pollak, MN; Schernhammer, ES; Hankinson, SE. Insulin-like growth factors and neoplasia. Nat. Rev. Cancer 2004, 4, 505–518. [Google Scholar] [Green Version]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [Green Version]
- Olayioye, MA; Neve, RM; Lane, HA; Hynes, NE. The ErbB signaling network: receptor heterodimerization in development and cancer. Embo. J. 2000, 19, 3159–3167. [Google Scholar] [Green Version]
- Sebolt-Leopold, JS; Herrera, R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat. Rev. Cancer 2004, 4, 937–947. [Google Scholar] [Green Version]
- Vivanco, I; Sawyers, CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [Green Version]
- Mukhtar, H; Ahmad, N. Tea polyphenols: prevention of cancer and optimizing health. Am. J. Clin. Nutr. 2000, 71, 1698S–1702S, discussion 1703S–1704S. [Google Scholar] [Green Version]
- Valcic, S; Muders, A; Jacobsen, NE; Liebler, DC; Timmermann, BN. Antioxidant chemistry of green tea catechins. Identification of products of the reaction of (–)-epigallocatechin gallate with peroxyl radicals. Chem. Res. Toxicol. 1999, 12, 382–386. [Google Scholar] [Green Version]
- Cao, Y; Cao, R. Angiogenesis inhibited by drinking tea. Nature 1999, 398, 381. [Google Scholar] [Green Version]
- Rodriguez, SK; Guo, W; Liu, L; Band, MA; Paulson, EK; Meydani, M. Green tea catechin, epigallocatechin-3-gallate, inhibits vascular endothelial growth factor angiogenic signaling by disrupting the formation of a receptor complex. Int. J. Cancer 2006, 118, 1635–1644. [Google Scholar] [Green Version]
- Han, C. Screening of anticarcinogenic ingredients in tea polyphenols. Cancer Lett. 1997, 114, 153–158. [Google Scholar] [Green Version]
- Surh, YJ; Chun, KS; Cha, HH; Han, SS; Keum, YS; Park, KK; Lee, SS. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-kappa B activation. Mutat. Res. 2001, 480–481, 243–268. [Google Scholar] [Green Version]
- Aggarwal, BB; Shishodia, S; Sandur, SK; Pandey, MK; Sethi, G. Inflammation and cancer: How hot is the link? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar] [Green Version]
- Shirakami, Y; Shimizu, M; Tsurumi, H; Hara, Y; Tanaka, T; Moriwaki, H. EGCG and polyphenon E attenuate inflammation-related mouse colon carcinogenesis induced by AOM and DSS. Mol. Med. Rep. 2008, 1, 355–361. [Google Scholar] [Green Version]
- Ahn, WS; Yoo, J; Huh, SW; Kim, CK; Lee, JM; Namkoong, SE; Bae, SM; Lee, IP. Protective effects of green tea extracts (polyphenon E and EGCG) on human cervical lesions. Eur. J. Cancer. Prev. 2003, 12, 383–390. [Google Scholar] [Green Version]
- Bettuzzi, S; Brausi, M; Rizzi, F; Castagnetti, G; Peracchia, G; Corti, A. Chemoprevention of human prostate cancer by oral administration of green tea catechins in volunteers with high-grade prostate intraepithelial neoplasia: a preliminary report from a one-year proof-of-principle study. Cancer Res. 2006, 66, 1234–1240. [Google Scholar] [Green Version]
- Schlessinger, J. Common and distinct elements in cellular signaling via EGF and FGF receptors. Science 2004, 306, 1506–1507. [Google Scholar] [Green Version]
- Chang, L; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [Green Version]
- Johnson, GL; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [Green Version]
- Kennedy, SG; Wagner, AJ; Conzen, SD; Jordan, J; Bellacosa, A; Tsichlis, PN; Hay, N. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes. Dev. 1997, 11, 701–713. [Google Scholar] [Green Version]
- Datta, SR; Dudek, H; Tao, X; Masters, S; Fu, H; Gotoh, Y; Greenberg, ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [Green Version]
- Romashkova, JA; Makarov, SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature 1999, 401, 86–90. [Google Scholar] [Green Version]
- Eferl, R; Wagner, EF. AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [Green Version]
- Karin, M; Cao, Y; Greten, FR; Li, ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [Green Version]
- Liang, YC; Lin-shiau, SY; Chen, CF; Lin, JK. Suppression of extracellular signals and cell proliferation through EGF receptor binding by (–)-epigallocatechin gallate in human A431 epidermoid carcinoma cells. J. Cell. Biochem. 1997, 67, 55–65. [Google Scholar] [Green Version]
- Pianetti, S; Guo, S; Kavanagh, KT; Sonenshein, GE. Green tea polyphenol epigallocatechin-3 gallate inhibits Her-2/neu signaling, proliferation, and transformed phenotype of breast cancer cells. Cancer Res. 2002, 62, 652–655. [Google Scholar] [Green Version]
- Graus-Porta, D; Beerli, RR; Daly, JM; Hynes, NE. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. Embo. J. 1997, 16, 1647–1655. [Google Scholar] [Green Version]
- Adhami, VM; Siddiqui, IA; Ahmad, N; Gupta, S; Mukhtar, H. Oral consumption of green tea polyphenols inhibits insulin-like growth factor-I-induced signaling in an autochthonous mouse model of prostate cancer. Cancer Res. 2004, 64, 8715–8722. [Google Scholar] [Green Version]
- Ferrara, N; Gerber, HP; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [Green Version]
- Ferrara, N. Vascular endothelial growth factor as a target for anticancer therapy. Oncologist 2004, 9 Suppl. 1, 2–10. [Google Scholar] [Green Version]
- Neuhaus, T; Pabst, S; Stier, S; Weber, AA; Schror, K; Sachinidis, A; Vetter, H; Ko, YD. Inhibition of the vascular-endothelial growth factor-induced intracellular signaling and mitogenesis of human endothelial cells by epigallocatechin-3 gallate. Eur. J. Pharmacol. 2004, 483, 223–227. [Google Scholar] [Green Version]
- Sah, JF; Balasubramanian, S; Eckert, RL; Rorke, EA. Epigallocatechin-3-gallate inhibits epidermal growth factor receptor signaling pathway. Evidence for direct inhibition of ERK1/2 and AKT kinases. J. Biol. Chem. 2004, 279, 12755–12762. [Google Scholar] [Green Version]
- Chung, JY; Park, JO; Phyu, H; Dong, Z; Yang, CS. Mechanisms of inhibition of the Ras-MAP kinase signaling pathway in 30.7b Ras 12 cells by tea polyphenols (–)-epigallocatechin-3-gallate and theaflavin-3,3′-digallate. Faseb. J. 2001, 15, 2022–2024. [Google Scholar] [Green Version]
- Dong, Z; Ma, W; Huang, C; Yang, CS. Inhibition of tumor promoter-induced activator protein 1 activation and cell transformation by tea polyphenols, (–)-epigallocatechin gallate, and theaflavins. Cancer Res. 1997, 57, 4414–4419. [Google Scholar] [Green Version]
- Nomura, M; Ma, W; Chen, N; Bode, AM; Dong, Z. Inhibition of 12-O-tetradecanoylphorbol-13-acetate-induced NF-kappaB activation by tea polyphenols, (–)-epigallocatechin gallate and theaflavins. Carcinogenesis 2000, 21, 1885–1890. [Google Scholar] [Green Version]
- Afaq, F; Adhami, VM; Ahmad, N; Mukhtar, H. Inhibition of ultraviolet B-mediated activation of nuclear factor kappaB in normal human epidermal keratinocytes by green tea Constituent (–)-epigallocatechin-3-gallate. Oncogene. 2003, 22, 1035–1044. [Google Scholar] [Green Version]
- Albanese, C; Johnson, J; Watanabe, G; Eklund, N; Vu, D; Arnold, A; Pestell, RG. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J. Biol. Chem. 1995, 270, 23589–23597. [Google Scholar] [Green Version]
- Guttridge, DC; Albanese, C; Reuther, JY; Pestell, RG; Baldwin, AS, Jr. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell. Biol. 1999, 19, 5785–5799. [Google Scholar] [Green Version]
- Tanabe, T; Tohnai, N. Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins Other Lipid Mediat. 2002, 68–69, 95–114. [Google Scholar] [Green Version]
- Yu, Q; Geng, Y; Sicinski, P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001, 411, 1017–1021. [Google Scholar] [Green Version]
- Pianetti, S; Arsura, M; Romieu-Mourez, R; Coffey, RJ; Sonenshein, GE. Her-2/neu overexpression induces NF-kappaB via a PI3-kinase/Akt pathway involving calpain-mediated degradation of IkappaB-alpha that can be inhibited by the tumor suppressor PTEN. Oncogene. 2001, 20, 1287–1299. [Google Scholar] [Green Version]
- Finco, TS; Westwick, JK; Norris, JL; Beg, AA; Der, CJ; Baldwin, AS, Jr. Oncogenic Ha-Ras-induced signaling activates NF-kappaB transcriptional activity, which is required for cellular transformation. J. Biol. Chem. 1997, 272, 24113–24116. [Google Scholar] [Green Version]
- Tamura, M; Sebastian, S; Yang, S; Gurates, B; Ferrer, K; Sasano, H; Okamura, K; Bulun, SE. Up-regulation of cyclooxygenase-2 expression and prostaglandin synthesis in endometrial stromal cells by malignant endometrial epithelial cells. A paracrine effect mediated by prostaglandin E2 and nuclear factor-kappa B. J. Biol. Chem. 2002, 277, 26208–26216. [Google Scholar] [Green Version]
- Vadlamudi, R; Mandal, M; Adam, L; Steinbach, G; Mendelsohn, J; Kumar, R. Regulation of cyclooxygenase-2 pathway by HER2 receptor. Oncogene. 1999, 18, 305–314. [Google Scholar] [Green Version]
- Sheng, H; Shao, J; Washington, MK; DuBois, RN. Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J. Biol. Chem. 2001, 276, 18075–18081. [Google Scholar] [Green Version]
- Sachinidis, A; Seul, C; Seewald, S; Ahn, H; Ko, Y; Vetter, H. Green tea compounds inhibit tyrosine phosphorylation of PDGF beta-receptor and transformation of A172 human glioblastoma. FEBS Lett. 2000, 471, 51–55. [Google Scholar] [Green Version]
- Tachibana, H; Koga, K; Fujimura, Y; Yamada, K. A receptor for green tea polyphenol EGCG. Nat. Struct. Mol. Biol. 2004, 11, 380–381. [Google Scholar] [Green Version]
- Fujimura, Y; Yamada, K; Tachibana, H. A lipid raft-associated 67kDa laminin receptor mediates suppressive effect of epigallocatechin-3-O-gallate on FcepsilonRI expression. Biochem. Biophys. Res. Commun. 2005, 336, 674–681. [Google Scholar] [Green Version]
- Tachibana, H; Fujimura, Y; Yamada, K. Tea polyphenol epigallocatechin-3-gallate associates with plasma membrane lipid rafts: lipid rafts mediate anti-allergic action of the catechin. Biofactors 2004, 21, 383–385. [Google Scholar] [Green Version]
- Pike, LJ; Casey, L. Cholesterol levels modulate EGF receptor-mediated signaling by altering receptor function and trafficking. Biochemistry 2002, 41, 10315–10322. [Google Scholar] [Green Version]
- Sottocornola, E; Misasi, R; Mattei, V; Ciarlo, L; Gradini, R; Garofalo, T; Berra, B; Colombo, I; Sorice, M. Role of gangliosides in the association of ErbB2 with lipid rafts in mammary epithelial HC11 cells. FEBS J. 2006, 273, 1821–1830. [Google Scholar] [Green Version]
- Remacle-Bonnet, M; Garrouste, F; Baillat, G; Andre, F; Marvaldi, J; Pommier, G. Membrane rafts segregate pro- from anti-apoptotic insulin-like growth factor-I receptor signaling in colon carcinoma cells stimulated by members of the tumor necrosis factor superfamily. Am. J. Pathol. 2005, 167, 761–773. [Google Scholar] [Green Version]
- Furuchi, T; Anderson, RG. Cholesterol depletion of caveolae causes hyperactivation of extracellular signal-related kinase (ERK). J. Biol. Chem. 1998, 273, 21099–21104. [Google Scholar] [Green Version]
- Roepstorff, K; Thomsen, P; Sandvig, K; van Deurs, B. Sequestration of epidermal growth factor receptors in non-caveolar lipid rafts inhibits ligand binding. J. Biol. Chem. 2002, 277, 18954–18960. [Google Scholar] [Green Version]
- Weinstein, IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science 2002, 297, 63–64. [Google Scholar] [Green Version]
- Ahmad, N; Gupta, S; Mukhtar, H. Green tea polyphenol epigallocatechin-3-gallate differentially modulates nuclear factor kappaB in cancer cells versus normal cells. Arch. Biochem. Biophys. 2000, 376, 338–346. [Google Scholar] [Green Version]
- Chow, HH; Cai, Y; Hakim, IA; Crowell, JA; Shahi, F; Brooks, CA; Dorr, RT; Hara, Y; Alberts, DS. Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals. Clin. Cancer Res. 2003, 9, 3312–3319. [Google Scholar] [Green Version]
- Bettuzzi, S; Brausi, M; Rizzi, F; Castagnetti, G; Peracchia, G; Corti, A. Chemoprevention of human prostate cancer by oral administration of green tea catechins in volunteers with high-grade prostate intraepithelial neoplasia: a preliminary report from a one-year proof-of-principle study. Cancer Res. 2006, 66, 1234–1240. [Google Scholar] [Green Version]
Share and Cite
Shimizu, M.; Shirakami, Y.; Moriwaki, H. Targeting Receptor Tyrosine Kinases for Chemoprevention by Green Tea Catechin, EGCG. Int. J. Mol. Sci. 2008, 9, 1034-1049. https://doi.org/10.3390/ijms9061034
Shimizu M, Shirakami Y, Moriwaki H. Targeting Receptor Tyrosine Kinases for Chemoprevention by Green Tea Catechin, EGCG. International Journal of Molecular Sciences. 2008; 9(6):1034-1049. https://doi.org/10.3390/ijms9061034
Chicago/Turabian StyleShimizu, Masahito, Yohei Shirakami, and Hisataka Moriwaki. 2008. "Targeting Receptor Tyrosine Kinases for Chemoprevention by Green Tea Catechin, EGCG" International Journal of Molecular Sciences 9, no. 6: 1034-1049. https://doi.org/10.3390/ijms9061034
APA StyleShimizu, M., Shirakami, Y., & Moriwaki, H. (2008). Targeting Receptor Tyrosine Kinases for Chemoprevention by Green Tea Catechin, EGCG. International Journal of Molecular Sciences, 9(6), 1034-1049. https://doi.org/10.3390/ijms9061034