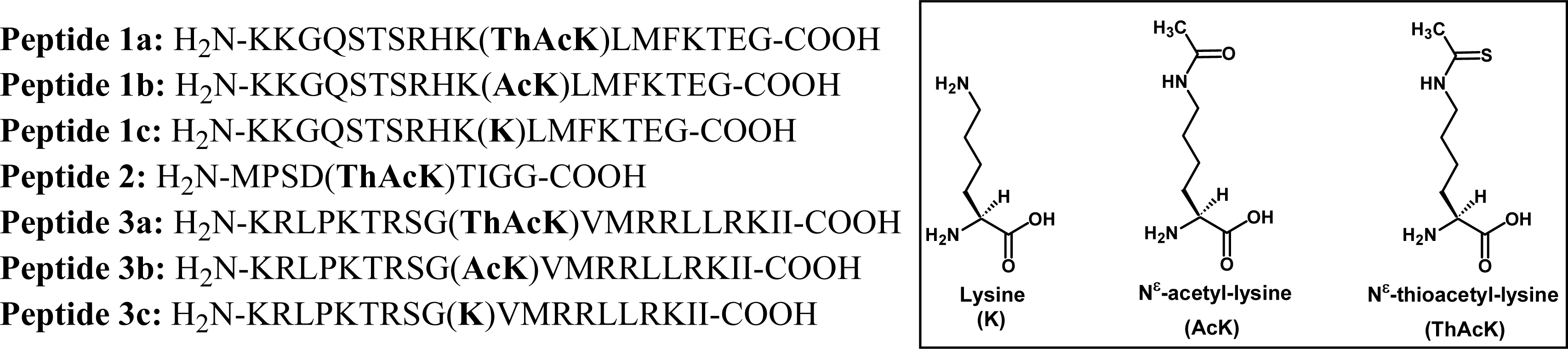Abstract
Inhibitors of human NAD+-dependent protein deacetylases possess great value for deciphering the biology of these enzymes and as potential therapeutics for metabolic and age-related diseases and cancer. In the current study, we have experimentally demonstrated that, the potent inhibition we obtained previously for one of these enzymes (i.e. sirtuin type 1 (SIRT1)) by simply replacing Nε-thioacetyl-lysine for Nε-acetyl-lysine in its peptide substrate, represented a general and efficient strategy to develop potent and selective inhibitors of human NAD+-dependent protein deacetylase enzymes. Indeed, by using this simple inhibition strategy, potent (low-micromolar) and selective (≤40-fold) SIRT2 and SIRT3 inhibitors, which were either comparable or superior to currently existing inhibitors, have also been quickly identified in the current study. These inhibitors could be used as chemical biological tools or as lead compounds for further focused structure-activity optimization.
1. Introduction
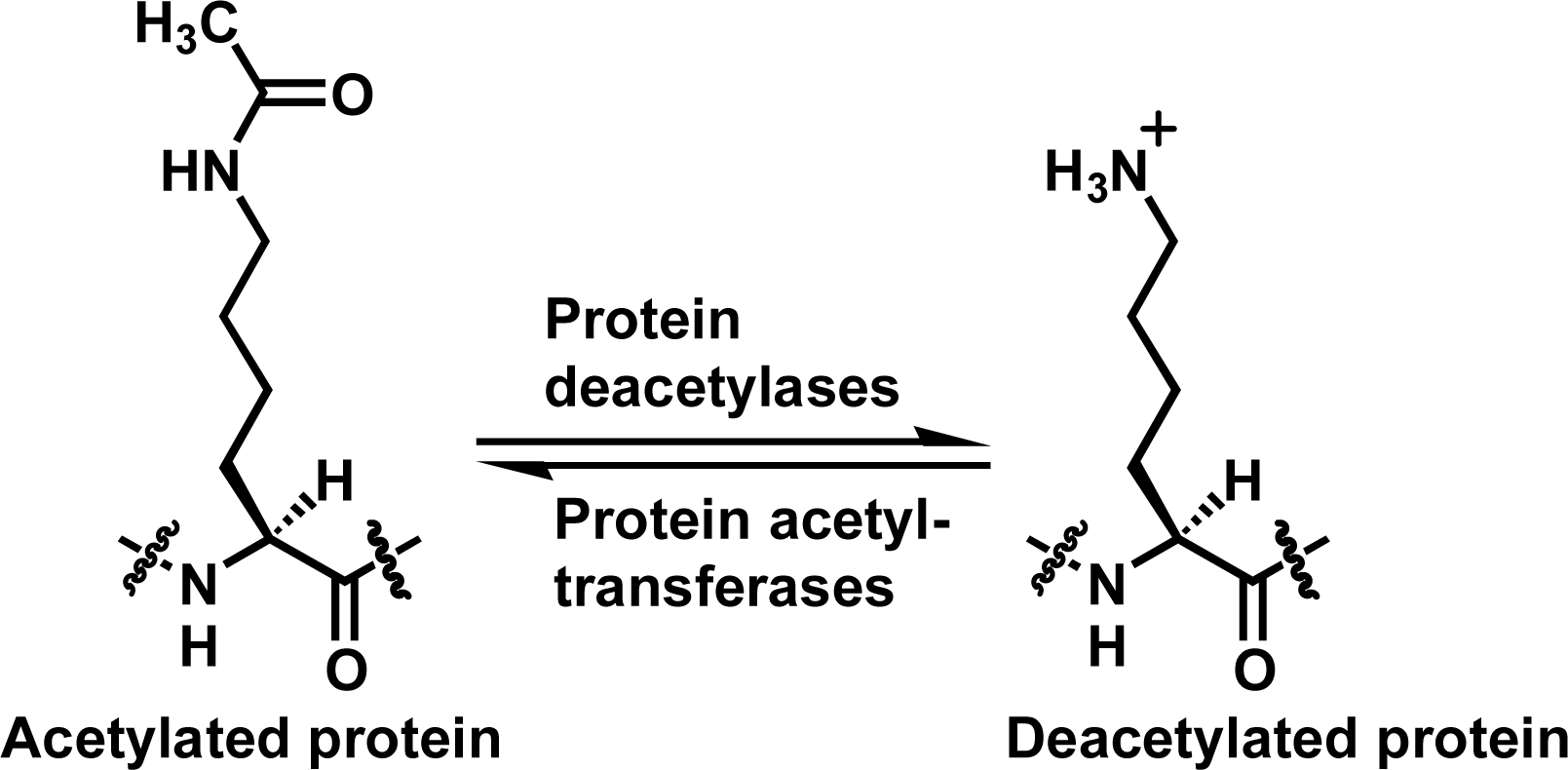
Reversible lysine Nε-acetylation on proteins has been increasingly recognized to play critical roles in regulating multiple pivotal cellular processes such as gene transcription, cell-cycle progression, and metabolism [1–13]. This reversible lysine Nε-acetylation is coordinated by protein acetyltransferases and protein deacetylases (Figure 1). As such, chemical modulation of these enzymes could offer therapeutic benefits for treating human diseases including metabolic and age-related diseases and cancer. This modulation could also be a chemical biological approach to further deciphering the biology of these enzymes.
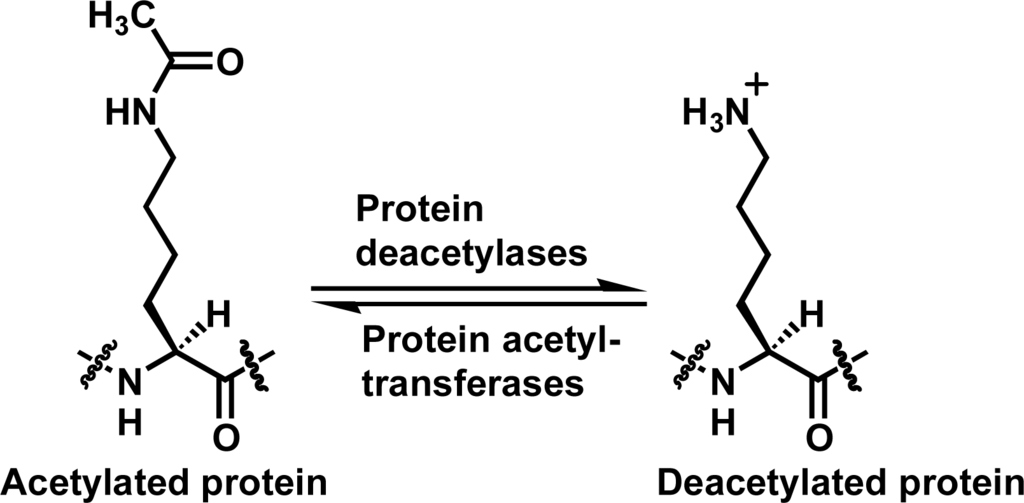
Figure 1.
The lysine Nε-acetylation and deacetylation reactions catalyzed respectively by protein acetyltransferases and protein deacetylases.
Human protein deacetylases have been categorized into class I, II, III, and IV subfamilies [6, 14, 15]. Class I, II, and IV (also collectively known as classical) enzymes include the 11 HDACs, i.e. HDAC1-11 (HDAC is the abbreviation for histone deacetylase that is named after the first discovered protein substrate histone), and they all require a catalytic zinc (Zn2+). Class III enzymes include the 7 human sirtuins, i.e. SIRT1-7 (sirtuin type 1–7). However, among the 7 human sirtuins, only SIRT1, SIRT2, SIRT3, and SIRT5 have been demonstrated to be bona fide protein deacetylases, and they all require coenzyme nicotinamide adenine dinucleotide (NAD+) for activity. SIRT4 and SIRT6 are ADPribosyltransferases, whereas an enzymatic activity for SIRT7 has not been identified.
As compared to the long-standing active pursuit of inhibitors of Zn2+-dependent classical enzymes, the development of inhibitors/activators of the NAD+-dependent protein deacetylases has only gained pace recently [8–13]. Regarding the inhibitor development for these latter enzymes, up to this date, there have been only two research reports disclosing the discovery of not only potent but also selective inhibitors, i.e. an indole-based SIRT1 inhibitor (IC50 ∼98 nM) reported by Napper, et al. [16] and a SIRT2 inhibitor (IC50 ∼3.5 μM) reported by Outeiro, et al. [17]. Weak (micromolar level) and/or nonselective (inhibiting multiple deacetylases within the class III subfamily and/or also inhibiting enzymes outside of this subfamily) inhibition was observed for all the other currently reported inhibitors whose potency and selectivity have been sufficiently addressed. Therefore, developing novel inhibition strategies and inhibitors for human NAD+-dependent protein deacetylases is expected to constitute an active research area in years to come.
By evaluating human p53 tumor suppressor protein C-terminal peptides (amino acid residues 372–389) containing Nε-thioacetyl-lysine (peptide 1a) or Nε-acetyl-lysine (Peptide 1b) at the 382 position (Figure 2), we previously demonstrated that, whereas the two peptides were comparably de(thio)acetylated by HDAC8, peptide 1a was a potent inhibitor for SIRT1 with an IC50 ∼2 μM [18], only secondary to the indole-based SIRT1 inhibitor reported by Napper, et al. [16]. Our previous experimental data further suggested that the observed potent SIRT1 inhibition by peptide 1a could be conferred by its processing by SIRT1, but with the formation of a longer-lived catalytically less competent intermediate following the nicotinamide cleavage step, as compared to the normal processing of peptide 1b by SIRT1 [18]. Because the catalytic domains were predicted to be highly conserved among the four currently known human NAD+-dependent protein deacetylases, i.e. SIRT1, SIRT2, SIRT3, and SIRT5 [14, 15, 19], we hypothesized that the potent SIRT1 inhibition by peptide 1a has defined a simple yet general and effective inhibition strategy for all human NAD+-dependent protein deacetylases, i.e. the transformation of peptide substrates to potent peptide inhibitors by simple replacement of Nε-thioacetyl-lysine for Nε-acetyl-lysine. Because physiological substrates have been identified for SIRT1, SIRT2, and SIRT3, but not yet for SIRT5 [6], we chose SIRT2 and SIRT3 to test our hypothesis.
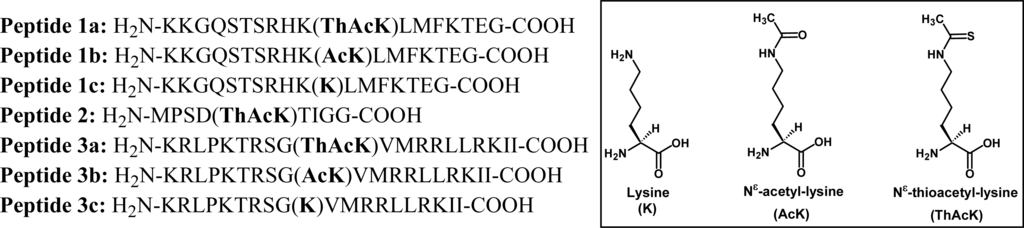
Figure 2.
Peptides used in the current study. The following peptide templates were used: Peptides 1a–c, SIRT1 substrate human p53 tumor suppressor protein (372–389) [18]; Peptide 2, SIRT2 substrate human α-tubulin (36–44); Peptides 3a–c, SIRT3 substrate human Acetyl-coenzyme A synthetase 2 (AceCS2) (633–652).
2. Results and Discussion
2.1 Peptide-based potent and selective inhibitors of SIRT1, SIRT2, and SIRT3
Figure 2 shows the amino acid sequences for all the peptides that were used in the current study. These peptides include peptides 1a-c that were also used previously by us [18, 20] and others [21–23] for various studies; peptide 2 that was based on the template derived from the SIRT2 substrate human α-tubulin; and peptides 3a-c that were based on the template derived from the SIRT3 substrate human AceCS2. Fmoc-chemistry-based strategy was employed for solid phase peptide synthesis [24], and Nα-Fmoc-Nε-thioacetyl-lysine [18] was used to incorporate Nε-thioacetyl-lysine into peptides.
Peptides 1b and 1c were used, respectively, as the substrate and the synthetic authentic deacetylation peptide product for the SIRT1 inhibition assay based on high pressure liquid chromatography (HPLC) [18]. Peptides 3b and 3c were used, respectively, as the substrate and the synthetic authentic deacetylation peptide product for the HPLC-based SIRT3 inhibition assay. Peptide 1b was also employed as an in vitro substrate in our HPLC-based SIRT2 inhibition assay because we found that, albeit being ∼8-fold less efficiently processed by SIRT2 as compared to by SIRT1, peptide 1b still gave rise to reliable signal when longer reaction times were used.
All the inhibition assays with SIRT1, SIRT2, and SIRT3 were performed under initial conditions (turnover of the limiting substrate was maintained at ≤12%) (see Experimental Section). Linear Dixon plots (1/v0 vs. [inhibitor]) were obtained from all the assay data, and were used to estimate IC50 values as an indication of the inhibition potency (Table 1).

Table 1.
Human sirtuin inhibitor evaluationa
It is apparent from Table 1 that peptides 2 and 3a also turned out to be potent inhibitors for SIRT2 (IC50 ∼11 μM) and SIRT3 (IC50 ∼5 μM), respectively. Indeed, peptide 3a represented the very first potent SIRT3 inhibitor ever identified. These data strongly supported our hypothesis that the potent SIRT1 inhibition by peptide 1a has defined a simple yet general and effective inhibition strategy for all human NAD+-dependent protein deacetylases, i.e. the transformation of peptide substrates to potent peptide inhibitors by simple replacement of Nε-thioacetyl-lysine for Nε-acetyl-lysine.
Peptides 1a, 2, and 3a were further evaluated for their possible selective inhibition among SIRT1, SIRT2, and SIRT3. As shown in Table 1: i) peptide 1a was found to have a comparable inhibition potency against SIRT2 to that against SIRT1, but a ∼35-fold weaker inhibition potency against SIRT3; ii) peptide 2 was found to be a ∼10-fold and ∼40-fold weaker inhibitor for SIRT1 and SIRT3, respectively, as compared to its inhibition against SIRT2; iii) peptide 3a was found to be a comparably potent inhibitor for both SIRT2 and SIRT3, but a ∼5-fold stronger inhibitor for SIRT1. The rationale behind our decision to evaluate the relative inhibition potencies of peptides 1a, 2, and 3a among SIRT1, SIRT2, and SIRT3 was that, if the amino acid residues surrounding the “warhead” (i.e. Nε-thioacetyl-lysine) in these peptides defined different “addresses” that were to be recognized by different enzymes, selective inhibition ought to be obtained. That we indeed observed different degrees of selective inhibition supported this rationale. However, several findings in our current study need further discussion, most notably the strong inhibition of peptide 1a against SIRT2 and peptide 3a against both SIRT1 and SIRT2 (Table 1), because peptides 1a and 3a were based on peptide templates derived from the SIRT1 physiological substrate human p53 protein and the SIRT3 physiological substrate human AceCS2, respectively. These results could imply that, under certain in vivo conditions, SIRT2 could also accept human p53 protein and AceCS2 as its substrates and SIRT1 could also accept AceCS2 as its substrate. Alternatively, these in vitro experimental data with purified recombinant enzymes may not fully account for the substrate selection by these enzymes in vivo, which could also be regulated by both spatial and temporal mechanisms.
2.2 Possible HDAC8-catalyzed dethioacetylation of peptide-based inhibitors
Our previous demonstration that peptides 1a and 1b were comparably de(thio)acetylated by HDAC8 [18] suggested that, when placed within an appropriate amino acid sequence, the thioacetyl group can serve as a functional mimic for the acetyl group for the enzymatic deacetylation reaction catalyzed by HDAC8. In a subsequent set of experiments employing the HeLa nuclear extracts enriched in HDAC1 and HDAC2 as well as the purified recombinant HDAC1 and HDAC2 as enzyme preparations, we previously further demonstrated that the replacement of Nε-thioacetyl-lysine for Nε-acetyl-lysine conferred resistance to HDACs other than HDAC8 [25]. Taken together, these previous experimental results argued for the notion that, when placed within an appropriate amino acid sequence, the thioacetyl group can serve as a functional mimic for the acetyl group only for HDAC8-catalyzed reaction. Therefore, in the current study, we sought to determine if peptides 2 and 3a could also be dethioacetylated by HDAC8, because the lability of a Nε-thioacetyl-lysine-containing peptide toward HDAC8 is expected to diminish its value as a chemical biological research tool or a potential therapeutic agent.
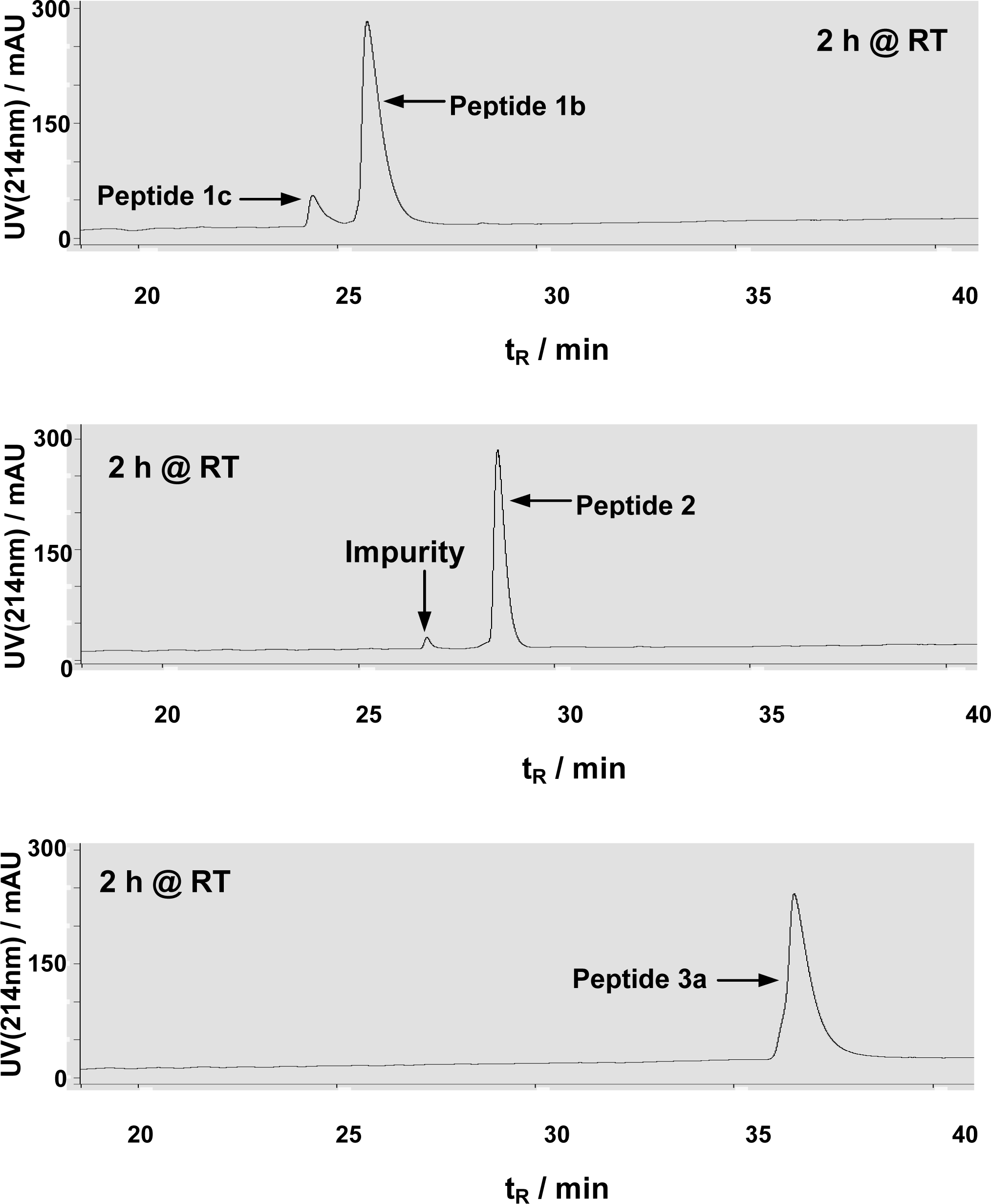
In a HPLC-based HDAC8 assay [18, 20], peptides 1b, 2, and 3a were allowed to be incubated for 2 h at room temperature in the HDAC8 assay buffer. While ∼10% substrate turnover from peptide 1b was observed, no detectable formation of the dethioacetylated peptide products was observed from both peptides 2 and 3a (Figure 3), indicating that, unlike peptides 1a and 1b, peptides 2 and 3a could not be dethioacetylated by HDAC8 under the same experimental conditions.
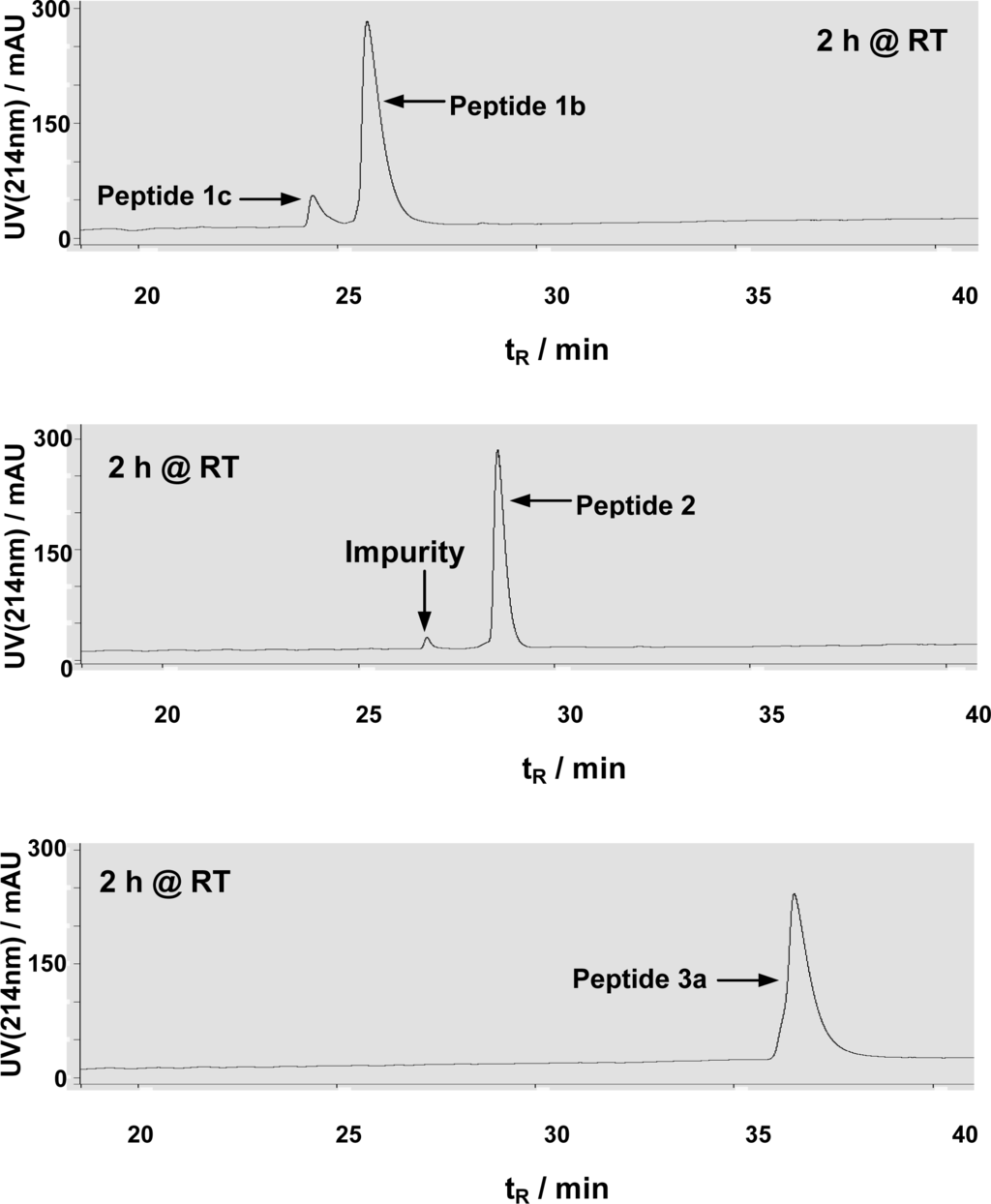
Figure 3.
Representative HPLC chromatograms from HDAC8 assays with peptides 1b, 2, and 3a. All assays were performed in duplicate and essentially the same HPLC chromatograms were obtained for duplicates. The small peak with tR∼27 min in the second chromatogram was from a minor impurity in the purified peptide 2 sample, rather than the dethioacetylated product. The lack of detectable dethioacetylation of peptide 3a was apparent from the absence of a peak with tR∼34 min (for peptide 3c) in the third chromatogram.
Taken together with section 2.1, we have experimentally demonstrated that, the potent inhibition we obtained previously for SIRT1 by simply replacing Nε-thioacetyl-lysine for Nε-acetyl-lysine in its peptide substrate, represented a general and efficient strategy to develop potent and selective inhibitors of human NAD+-dependent protein deacetylase enzymes. Furthermore, we have identified a potent and selective SIRT2 inhibitor (peptide 2) and a potent pan-sirtuin (among SIRT1, SIRT2, and SIRT3) inhibitor (peptide 3a) that showed resistance to enzymatic dethioacetylation by classical HDAC enzymes. Peptides 2 and 3a will thus be expected to be valuable as chemical tools for deciphering the biology of these human sirtuins. Even though these peptide-based inhibitors might not be cell permeable, they could be ferried inside a cell by their conjugation (e.g. via a disulfide linkage) to various types of protein transduction domain (PTD) peptides [26–30]. Once a PTD peptide carries an inhibitor as cargo across cellular membranes, the cargo can be released inside a cell (e.g. following the cleavage of a disulfide linkage due to a reductive intracellular environment). Peptides 2 and 3a shall also be valuable lead compounds for developing inhibitors with enhanced potency, selectivity, metabolic stability, and cellular membrane permeability. Toward this goal of developing improved inhibitors suitable for in vivo applications, the following stepwise strategy could be considered. i) To obtain structurally more manageable minimal peptide sequences via peptide truncation. The feasibility of peptide sequence truncation without drastic loss of inhibition potency could be supported by the following studies [31, 32]: a) a previous X-ray structural analysis of a sirtuin enzyme with peptide 1b showed that Nε-acetyl-lysine and the two amino acid residues on each side of Nε-acetyl-lysine were the peptide residues that made predominant binding interactions with the sirtuin enzyme used; b) multiple 5-mer peptides harboring Nε-acetyl-lysine in their middle positions were identified from a combinatorial peptide library to serve as good SIRT1 substrates. This feasibility was further directly supported by our observed modest decrease (ca. 6-fold) in inhibition potency following truncation of the 18-mer peptide 1a to a 5-mer peptide (i.e. H2N-HK(ThAcK)LM-COOH) (Fatkins and Zheng, unpublished results). ii) To construct and screen (against various human sirtuins) a focused peptide library in which all the library members will have Nε-thioacetyl-lysine occupying their middle positions. iii) To perform further medicinal chemistry manipulations on library hits. While maintaining the potency and selectivity of hits, their metabolic stability and cellular membrane permeability could be potentially enhanced through the minimization of their peptidic nature.
3. Experimental Section
3.1 Peptide synthesis and purification
All peptides were synthesized based on the Fmoc chemistry strategy on a PS3 peptide synthesizer (Protein Technologies Inc., Tucson, AZ, USA). Except Nα-Fmoc-Nε-thioacetyl-lysine, all other Fmocprotected amino acids and pre-loaded Wang resins were purchased from Novabiochem (La Jolla, CA, USA). Nα-Fmoc-Nε-thioacetyl-lysine was synthesized from Nα-Fmoc-lysine and ethyl dithioacetate as described previously [18]. For each coupling reaction, 4 equivalents of a Fmoc-protected amino acid, 3.8–4.0 equivalents of the coupling reagent 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HBTU) and the additive N-hydroxybenzotriazole (HOBt) were used in the presence of 0.4 M 4-methylmorpholine (NMM)/DMF, and the coupling reaction was allowed to proceed at room temperature for 1 h. A 20% (v/v) piperidine/DMF solution was used for Fmoc removal. All the peptides were cleaved from the resins by reagent K (83.6% (v/v) trifluoroacetic acid, 5.9% (v/v) phenol, 4.2% (v/v) double-deionized water (ddH2O), 4.2% (v/v) thioanisole, 2.1% (v/v) ethanedithiol), precipitated in cold diethyl ether, and purified by reversed-phase HPLC on a preparative C18 column (100 Å, 2.14 x 25 cm). The column was eluted with a gradient of ddH2O containing 0.05% (v/v) of trifluoroacetic acid and acetonitrile containing 0.05% (v/v) of trifluoroacetic acid at 10 mL/min and monitored at 214 nm. The pooled HPLC fractions were stripped of acetonitrile and lyophilized to give all peptides as puffy white solids. Peptide purity (>95%) was verified by reversed-phase HPLC on an analytical C18 column (100 Å, 0.46 x 25 cm). The column was eluted with a gradient of ddH2O containing 0.05% (v/v) of trifluoroacetic acid and acetonitrile containing 0.05% (v/v) of trifluoroacetic acid at 1 mL/min and monitored at 214 nm. The molecular weights of all purified peptides were confirmed by matrix assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometric analysis. Peptide 1a: 2149 [M+H]+; Peptide 1b: 2133 [M+H]+; Peptide 1c: 2091 [M+H]+; Peptide 2: 964 [M+H]+; Peptide 3a: 2508 [M+H]+; Peptide 3b: 2492 [M+H]+; Peptide 3c: 2450 [M+H]+.
3.2 Inhibition assays with recombinant SIRT1, SIRT2, and SIRT3
GST-SIRT1 (whose plasmid is a kind gift from Prof. Tony Kouzarides) that we expressed and purified from Escherichia coli according to literature protocols [33–35] was used for SIRT1 inhibition assay since we have previously demonstrated the comparable activities of GST-free SIRT1 and GSTSIRT1 toward both peptides 1a and 1b [18]. For SIRT2 and SIRT3 inhibition assays, we used the tagfree SIRT2 (Cat. # SE251-0500) and the tag-free SIRT3 (Cat. #SE270-0500) from BIOMOL International L.P. (Plymouth Meeting, PA, USA). Essentially the same procedures were used for the inhibition assays with SIRT1, SIRT2, and SIRT3 enzymes, and were performed as described previously for the HPLC-based SIRT1 assay [18, 20]. In brief, an inhibition assay solution had the following components: 25 mM (or 50 mM for SIRT2 assay) Tris•HCl (pH 8.0), 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1 mg/mL BSA (SIGMA Cat. #A3803 with reduced fatty acid content, for SIRT2 assay only), 0.5 mM β-NAD+, 0.3 mM peptide substrate (peptide 1b for SIRT1 and SIRT2 assays; peptide 3b for SIRT3 assay), an inhibitor (peptide 1a, 2, or 3a) with varied concentrations including 0, and an enzyme (GST-SIRT1, 0.15 μM; SIRT2, 0.3 μM; or SIRT3, 2.0 μM). An enzymatic reaction was initiated by the addition of an enzyme at 37 °C and was allowed to be incubated at 37 °C for 10 min (for SIRT1 assay) or 60 min (for SIRT2 and SIRT3 assays) until quenched with the following stop solution: 100 mM HCl and 0.16 M acetic acid. Turnover of the limiting substrate was maintained at ≤12%. The quenched assay solutions were directly injected into a reversed-phase HPLC C18 column (100 Å, 0.46 x 25 cm), eluting with the following gradients of ddH2O containing 0.05% (v/v) trifluoroacetic acid (mobile phase A) and acetonitrile containing 0.05% (v/v) trifluoroacetic acid (mobile phase B): linear increase from 0% B to 35% B (for SIRT1 assay) or 40% B (for SIRT2 and SIRT3 assays) from 0–40 min (1 mL/min), and UV monitoring at 214 nm. The enzymatic deacetylation products (peptide 1c in SIRT1 and SIRT2 assays; peptide 3c in SIRT3 assay) were confirmed by their comigration with the chemically synthesized authentic samples and by MALDITOF mass spectrometric analysis, and were quantified by HPLC peak integration and comparison with those of synthetic authentic samples. Under the same assay conditions, no detectable formation of the deacetylation products (i.e. peptides 1c and 3c) was observed for non-enzymatic reactions. Furthermore, under the same assay conditions, peptides 1a and 3a did not give rise to detectable formation of peptides 1c and 3c, respectively, via dethioacetylation. No detectable formation of the corresponding dethioacetylated peptide from peptide 2 was also observed under the same assay conditions. All peptide stock solutions were prepared in ddH2O. IC50 values were estimated from Dixon plots (1/v0 vs. [inhibitor]) [36] as an indication of the inhibition potency.
3.3 Assay with recombinant HDAC8
The rcombinant HDAC8 (Cat. #SE145-0100) was purchased from BIOMOL International L.P. (Plymouth Meeting, PA, USA). The HPLC-based HDAC8 assay was performed as previously described [18, 20]. In brief, a HDAC8 assay solution had the following components: 25 mM Tris•HCl (pH 8.0), 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1 mg/mL BSA (SIGMA Cat. #A3803 with reduced fatty acid content), 0.3 mM peptide 1b, 2, or 3a, and 1.5 μM HDAC8. An enzymatic reaction was initiated by the addition of HDAC8 at room temperature and was allowed to be incubated at room temperature for 2 h before quenched with the following stop solution: 1.0 M HCl and 0.16 M acetic acid. The quenched assay solutions were directly injected into a reversed-phase HPLC C18 column (100 Å, 0.46 x 25 cm), eluting with the following gradients of ddH2O containing 0.05% (v/v) trifluoroacetic acid (mobile phase A) and acetonitrile containing 0.05% (v/v) trifluoroacetic acid (mobile phase B): linear increase from 0% B to 35% B (for the assay with peptide 2 or 1b) or 40% B (for the assay with peptide 3a or 1b) from 0–40 min (1 mL/min), and UV monitoring at 214 nm. Under the HDAC8 assay conditions, the deacetylated peptide (i.e. peptide 1c) was formed from peptide 1b as we observed previously [18, 20], however, no detectable formation of the corresponding dethioacetylated peptides from peptides 2 and 3a was observed. The enzymatically formed peptide 1c was confirmed by its comigration with the chemically synthesized authentic sample and by MALDITOF mass spectrometric analysis, and was quantified by HPLC peak integration and comparison with that of synthetic authentic sample. Under the same assay conditions, no detectable formation of peptide 1c was observed for non-enzymatic reactions.
4. Conclusions
In the current study, we have experimentally demonstrated that, the potent inhibition we obtained previously for SIRT1 by simply replacing Nε-thioacetyl-lysine for Nε-acetyl-lysine in its peptide substrate, represented a general and efficient strategy to develop potent and selective inhibitors of human NAD+-dependent protein deacetylase enzymes. Due to their resistance to enzymatic dethioacetylation by classical HDAC enzymes, the potent and selective SIRT2 inhibitor peptide 2 and the potent pan-sirtuin (among SIRT1, SIRT2, and SIRT3) inhibitor peptide 3a identified in the current study will be expected to be valuable as chemical tools for deciphering the biology of these human sirtuins. The availability of various types of PTD peptides [26–30] should promote the cellular applications of these peptide-based inhibitors by carrying them through cellular membranes. Furthermore, peptides 2 and 3a shall be valuable lead compounds for further focused structure-activity optimization.
Acknowledgements
We thank the James L. and Martha J. Foght Endowment, the University of Akron Research Foundation, and the University of Akron Faculty Research Fellowship for financial support. We thank Prof. Tony Kouzarides (University of Cambridge, UK) for the GST-SIRT1 plasmid, and Prof. Chrys Wesdemiotis and his research group at the University of Akron for the assistance with mass spectrometric analysis for the peptides used in the current study
Notes Added in Proof
When this manuscript was under review, Smith, et al. described a detailed mechanistic study of the potent inhibition of a Nε-thioacetyl-lysine-containing human histone H3 peptide (H2NKSTGGKXAPRKQ-OH, wherein X is Nε-thioacetyl-lysine) against Hst2, a yeast sirtuin enzyme [37]. Their conclusion that this histone H3 peptide behaved as a mechanism-based inhibitor agreed with our previous mechanistic suggestion that the observed potent SIRT1 inhibition by peptide 1a could be conferred by its processing by SIRT1, but with the formation of a longer-lived catalytically less competent intermediate following the nicotinamide cleavage step, as compared to the normal processing of peptide 1b by SIRT1 [18]. In their paper [37], Smith, et al. also described their discovery that the above histone H3 peptide behaved as a potent inhibitor against SIRT1, SIRT2, and SIRT3 with respective IC50 values being 2.0 ± 0.2 μM, 5.6 ± 0.8 μM, and 2.3 ± 0.3 μM, when assayed against the sirtuin-catalyzed deacetylation of a 3H-acetylated histone H3 peptide substrate. However, whether or not their inhibitor could be dethioacetylated by HDAC8 was not examined.
References
- Yang, XJ; Seto, E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar]
- Glozak, MA; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar]
- Hodawadekar, SC; Marmorstein, R. Chemistry of acetyl transfer by histone modifying enzymes: structure, mechanism and implications for effector design. Oncogene 2007, 26, 5528–5540. [Google Scholar]
- Batta, K; Das, C; Gadad, S; Shandilya, J; Kundu, TK. Reversible acetylation of non histone proteins: role in cellular function and disease. Subcell Biochem 2007, 41, 193–212. [Google Scholar]
- Shahbazian, MD; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem 2007, 76, 75–100. [Google Scholar]
- Saunders, LR; Verdin, E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene 2007, 26, 5489–5504. [Google Scholar]
- Swaminathan, V; Reddy, BA; Ruthrotha Selvi, B; Sukanya, MS; Kundu, TK. Small molecule modulators in epigenetics: implications in gene expression and therapeutics. Subcell Biochem 2007, 41, 397–428. [Google Scholar]
- Marchion, D; Munster, P. Development of histone deacetylase inhibitors for cancer treatment. Expert Rev Anticancer Ther 2007, 7, 583–598. [Google Scholar]
- Xu, WS; Parmigiani, RB; Marks, PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar]
- Duvic, M; Vu, J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous Tcell lymphoma. Expert Opin Investig Drugs 2007, 16, 1111–1120. [Google Scholar]
- Guarente, L. Sirtuins as potential targets for metabolic syndrome. Nature 2006, 444, 868–874. [Google Scholar]
- Garske, AL; Smith, BC; Denu, JM. Linking SIRT2 to Parkinson's disease. ACS Chem Biol 2007, 2, 529–532. [Google Scholar]
- Gan, L. Therapeutic potential of sirtuin-activating compounds in Alzheimer's disease. Drug News Perspect 2007, 20, 233–239. [Google Scholar]
- Thiagalingam, S; Cheng, KH; Lee, HJ; Mineva, N; Thiagalingam, A; Ponte, JF. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci 2003, 983, 84–100. [Google Scholar]
- Gregoretti, IV; Lee, YM; Goodson, HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol 2004, 338, 17–31. [Google Scholar]
- Napper, AD; Hixon, J; McDonagh, T; Keavey, K; Pons, JF; Barker, J; Yau, WT; Amouzegh, P; Flegg, A; Hamelin, E; Thomas, RJ; Kates, M; Jones, S; Navia, MA; Saunders, JO; DiStefano, PS; Curtis, R. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J Med Chem 2005, 48, 8045–8054. [Google Scholar]
- Outeiro, TF; Kontopoulos, E; Altmann, SM; Kufareva, I; Strathearn, KE; Amore, AM; Volk, CB; Maxwell, MM; Rochet, JC; McLean, PJ; Young, AB; Abagyan, R; Feany, MB; Hyman, BT; Kazantsev, AG. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science 2007, 317, 516–519. [Google Scholar]
- Fatkins, DG; Monnot, AD; Zheng, W. Nε-thioacetyl-lysine: a multi-facet functional probe for enzymatic protein lysine Nε-deacetylation. Bioorg Med Chem Lett 2006, 16, 3651–3656. [Google Scholar]
- Denu, JM. The Sir 2 family of protein deacetylases. Curr Opin Chem Biol 2005, 9, 431–440. [Google Scholar]
- Jamonnak, N; Fatkins, DG; Wei, L; Zheng, W. N(epsilon)-Methanesulfonyl-lysine as a nonhydrolyzable functional surrogate for N(epsilon)-acetyl-lysine. Org Biomol Chem 2007, 5, 892–896. [Google Scholar]
- Sauve, AA; Celic, I; Avalos, J; Deng, H; Boeke, JD; Schramm, VL. Chemistry of gene silencing: the mechanism of NAD+-dependent deacetylation reactions. Biochemistry 2001, 40, 15456–15463. [Google Scholar]
- Smith, JS; Avalos, J; Celic, I; Muhammad, S; Wolberger, C; Boeke, JD. SIR2 family of NAD(+)-dependent protein deacetylases. Methods Enzymol 2002, 353, 282–300. [Google Scholar]
- Avalos, JL; Celic, I; Muhammad, S; Cosgrove, MS; Boeke, JD; Wolberger, C. Structure of a Sir2 enzyme bound to an acetylated p53 peptide. Mol Cell 2002, 10, 523–535. [Google Scholar]
- Wellings, DA; Atherton, E. Standard Fmoc protocols. Methods Enzymol 1997, 289, 44–67. [Google Scholar]
- Fatkins, DG; Zheng, W. A spectrophotometric assay for histone deacetylase 8. Anal Biochem 2008, 372, 82–88. [Google Scholar]
- Wadia, JS; Dowdy, SF. Transmembrane delivery of protein and peptide drugs by TATmediated transduction in the treatment of cancer. Adv Drug Deliv Rev 2005, 57, 579–596. [Google Scholar]
- Fuchs, SM; Raines, RT. Polyarginine as a multifunctional fusion tag. Protein Sci 2005, 14, 1538–1544. [Google Scholar]
- Rothbard, JB; Jessop, TC; Wender, PA. Adaptive translocation: the role of hydrogen bonding and membrane potential in the uptake of guanidinium-rich transporters into cells. Adv Drug Deliv Rev 2005, 57, 495–504. [Google Scholar]
- Wright, LR; Rothbard, JB; Wender, PA. Guanidinium rich peptide transporters and drug delivery. Curr Protein Pept Sci 2003, 4, 105–124. [Google Scholar]
- Zheng, Y; Balasubramanyam, K; Cebrat, M; Buck, D; Guidez, F; Zelent, A; Alani, RM; Cole, PA. Synthesis and evaluation of a potent and selective cell-permeable p300 histone acetyltransferase inhibitor. J Am Chem Soc 2005, 127, 17182–17183. [Google Scholar]
- Avalos, JL; Celic, I; Muhammad, S; Cosgrove, MS; Boeke, JD; Wolberger, C. Structure of a Sir2 enzyme bound to an acetylated p53 peptide. Mol Cell 2002, 10, 523–535. [Google Scholar]
- Garske, AL; Denu, JM. SIRT1 top 40 hits: use of one-bead, one-compound acetyl-peptide libraries and quantum dots to probe deacetylase specificity. Biochemistry 2006, 45, 94–101. [Google Scholar]
- Langley, E; Pearson, M; Faretta, M; Bauer, UM; Frye, RA; Minucci, S; Pelicci, PG; Kouzarides, T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J 2002, 21, 2383–2396. [Google Scholar]
- Bannister, AJ; Cook, A; Kouzarides, T. In vitro DNA binding activity of Fos/Jun and BZLF1 but not C/EBP is affected by redox changes. Oncogene 1991, 6, 1243–1250. [Google Scholar]
- North, BJ; Schwer, B; Ahuja, N; Marshall, B; Verdin, E. Preparation of enzymatically active recombinant class III protein deacetylases. Methods 2005, 36, 338–345. [Google Scholar]
- Dixon, M. The determination of enzyme inhibitor constants. Biochem J 1953, 55, 170–171. [Google Scholar]
- Smith, BC; Denu, JM. Mechanism-based inhibition of sir2 deacetylases by thioacetyl-lysine peptide. Biochemistry 2007, 46, 14478–14486, Published on Web 11/21/2007. [Google Scholar]