
Immune Checkpoint Signatures Reveal Stage-Specific Biomarkers for High-Activity Multiple Sclerosis

 , , , ,
, , , , 
Abstract
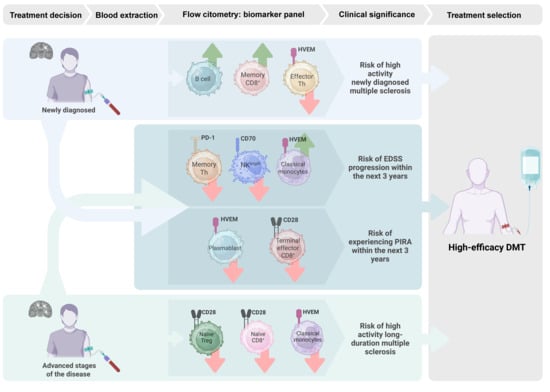
1. Introduction
2. Results
2.1. Unsupervised t-SNE Analysis Reveals Immune Cell Heterogeneity in Early-Stage MS Patients and Those with Long-Standing Disease
2.2. Immune Signatures Distinguish HAMS at Diagnosis in Early-Stage MS Patients
2.3. The Immunophenotypic Profile Identifies Therapeutic Resistance in Long-Standing MS and Recognizes Candidates for High-Efficacy Interventions
2.4. Immunophenotypic Markers Identify True Poor-Prognosis Patients
2.5. CD28 Expression on Terminal Effector CD8+ T Cells and HVEM in Plasmablasts as Predictors for Progression Independent of Relapse Activity
2.6. PD-1 Expression on Memory Th Cells as a Biomarker of True Prognosis of Disease Stability
3. Discussion
4. Materials and Methods
4.1. Study Design
- Newly diagnosed and treatment-naïve (early-stage MS patients): These patients were newly diagnosed and had not yet received any DMT at the time of sampling. Patients were further subdivided according to disease activity:
- 1.1.
- Early high-activity MS (e-HAMS): Males or females > 18 years recently diagnosed with MS who have not yet received DMT and exhibit the presence of at least two of the following poor prognostic factors: ≥20 lesions on MRI, ≥2 spinal/brainstem lesions, ≥2 gadolinium-enhancing lesions, ≥2 relapses per year, or EDSS ≥ 3.
- 1.2.
- Early non-high-activity MS (e-non HAMS): Males or females > 18 years who are recently diagnosed, treatment-naïve MS patients who have experienced one or fewer of the poor prognostic factors defined in the previous group.
- Long-standing MS patients (long-standing disease): Males or females over 18 years of age with MS who had received at least one prior DMT and had already completed the washout period required in the product information.
- 2.1.
- Long-standing high-activity MS (ls-HAMS): Males or females > 18 years with long-standing MS who had previously failed treatment, as evidenced by relapses, activity on MRI, or disability progression, despite prior therapy.
- 2.2.
- Long-standing non-high-activity MS (ls-non HAMS): Males or females > 18 years with long-standing MS who had previously received DMT and showed no signs of high disease activity at the time of inclusion. All patients had discontinued treatment for reasons other than therapeutic failure (risk of side effects, pregnancy, or patient choice).
4.2. Ethical Statement
4.3. Demographic and Clinical Data
4.4. Blood Sample Processing
4.5. Neurofilament Light Chain Quantification
4.6. Immunophenotypic Profiling of Immune Cell Subpopulations
4.7. Analysis of High Dimensional Data Using t-SNE
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CNS | central nervous system |
| DMTs | disease-modifying therapies |
| EDSS | expanded disability status scale |
| e-HAMS | early high-activity MS |
| e-non HAMS | early non-high-activity MS |
| HAMS | highly active multiple sclerosis |
| ICs | immune checkpoint |
| ls-HAMS | long-standing high-activity MS |
| ls-non HAMS | long-standing non-high-activity MS |
| MRI | magnetic resonance imaging |
| MS | multiple sclerosis |
| NfL | neurofilament light chain |
| PBMCs | peripheral blood mononuclear cells |
| PIRA | progression independent of relapse activity |
| PMS | progressive MS |
| PPMS | primary progressive MS |
| RAW | relapse associated worsening |
| ROC | receiver operating characteristic |
| RRMS | relapsing-remitting MS |
| SPMS | secondary progressive MS |
| t-SNE | T-distributed stochastic neighbor embedding |
| Th | T helper cell |
References
- Bierhansl, L.; Hartung, H.-P.; Aktas, O.; Ruck, T.; Roden, M.; Meuth, S.G. Thinking outside the box: Non-canonical targets in multiple sclerosis. Nat. Rev. Drug Discov. 2022, 21, 578–600. [Google Scholar] [CrossRef] [PubMed]
- Friese, M.A.; Schattling, B.; Fugger, L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 2014, 10, 225–238. [Google Scholar] [CrossRef]
- Kamińska, J.; Koper, O.M.; Piechal, K.; Kemona, H. Multiple sclerosis etiology and diagnostic potential. Postepy Hig. Med. Dosw. 2017, 71, 551–563. [Google Scholar] [CrossRef]
- Díaz, C.; Zarco, L.A.; Rivera, D.M. Highly active multiple sclerosis: An update. Mult. Scler. Relat. Disord. 2019, 30, 215–224. [Google Scholar] [CrossRef]
- Iacobaeus, E.; Arrambide, G.; Amato, M.P.; Derfuss, T.; Vukusic, S.; Hemmer, B.; Tintoré, M.; Brundin, L. Aggressive multiple sclerosis (1): Towards a definition of the phenotype. Mult. Scler. 2020, 26, 1352458520925369. [Google Scholar] [CrossRef] [PubMed]
- Meca-Lallana, J.; Yélamos, S.M.; Eichau, S.; Llaneza, M.; Martínez, J.M.; Martínez, J.P.; Lallana, V.M.; Torres, A.A.; Torres, E.M.; Río, J.; et al. Consensus statement of the Spanish Society of Neurology on the treatment of multiple sclerosis and holistic patient management in 2023. Neurologia 2024, 39, 196–208. [Google Scholar] [CrossRef]
- Meca-Lallana, J.; García-Merino, J.A.; Martínez-Yélamos, S.; Vidal-Jordana, A.; Costa, L.; Eichau, S.; Rovira, À.; Brieva, L.; Agüera, E.; Zarranz, A.R.-A. Identification of patients with relapsing multiple sclerosis eligible for high-efficacy therapies. Neurodegener. Dis. Manag. 2021, 11, 251–261. [Google Scholar] [CrossRef]
- Yang, J.; Hamade, M.; Wu, Q.; Wang, Q.; Axtell, R.; Giri, S.; Mao-Draayer, Y. Current and future biomarkers in multiple sclerosis. Int. J. Mol. Sci. 2022, 23, 5877. [Google Scholar] [CrossRef]
- Mehlhop-Williams, E.R.; Bevan, M.J. Memory CD8+ T cells exhibit increased antigen threshold requirements for recall proliferation. J. Exp. Med. 2014, 211, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Seals, M.R.; Moran, M.M.; Leavenworth, J.D.; Leavenworth, J.W. Contribution of dysregulated B-cells and IgE antibody responses to multiple sclerosis. Front. Immunol. 2022, 13, 900117. [Google Scholar] [CrossRef]
- del Rio, M.-L.; Fernandez-Renedo, C.; Scheu, S.; Pfeffer, K.; Shintani, Y.; Kronenberg, M.; Chaloin, O.; Schneider, P.; Rodriguez-Barbosa, J.-I. Therapeutic blockade of LIGHT interaction with herpesvirus entry mediator and lymphotoxin β receptor attenuates in vivo cytotoxic allogeneic responses. Transplantation 2014, 98, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- López-Molina, M.; Iglesias, G.T.; María-Diez, G.S.d.S.; Valentín-Quiroga, J.; Laso-García, F.; Gallego, R.; Pozo-Novoa, J.; Chamorro, B.; López-Collazo, E.; Puertas, I.; et al. Immune checkpoint-based biomarkers for therapeutic response in patients with multiple sclerosis. Front. Immunol. 2025, 16, 1694021. [Google Scholar] [CrossRef] [PubMed]
- Monreal, E.; Fernández-Velasco, J.I.; Álvarez-Lafuente, R.; de la Maza, S.S.; García-Sánchez, M.I.; Llufriu, S.; Casanova, B.; Comabella, M.; Martínez-Yélamos, S.; Galimberti, D.; et al. Serum biomarkers at disease onset for personalized therapy in multiple sclerosis. Brain 2024, 147, 4084–4093. [Google Scholar] [CrossRef] [PubMed]
- Bănică, L.; Vlaicu, O.; Jipa, R.; Abagiu, A.; Nicolae, I.; Neaga, E.; Oțelea, D.; Paraschiv, S. Exhaustion and senescence of CD4 and CD8 T cells that express co-stimulatory molecules CD27 and CD28 in subjects that acquired HIV by drug use or by sexual route. Germs 2021, 11, 66–77. [Google Scholar] [CrossRef]
- Shui, J.W.; Steinberg, M.W.; Kronenberg, M. Regulation of inflammation, autoimmunity, and infection immunity by HVEM-BTLA signaling. J. Leukoc. Biol. 2011, 89, 517–523. [Google Scholar] [CrossRef]
- Kim, W.; Bae, E.; Kang, Y.; Bae, H.; Hong, S.H.; Lee, J.Y.; Park, J.; Kwon, B.S.; Suk, K.; Lee, W. Glucocorticoid-induced tumour necrosis factor receptor family related protein (GITR) mediates inflammatory activation of macrophages that can destabilize atherosclerotic plaques. Immunology 2006, 119, 421–429. [Google Scholar] [CrossRef]
- Smolders, J.; Hamann, J. Programmed cell death protein 1-positive CD8+ T cells in multiple sclerosis: Exhausted fighters or peacekeepers. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1173. [Google Scholar] [CrossRef]
- Roe, K. Immunoregulatory natural killer cells. Clin. Chim. Acta 2024, 558, 117896. [Google Scholar] [CrossRef]
- Zhang, Q.; Vignali, D.A.A. Co-stimulatory and co-inhibitory pathways in autoimmunity. Immunity 2016, 44, 1034–1051. [Google Scholar] [CrossRef]
- Kang, Y.M.; Kim, S.Y.; Kang, J.H.; Han, S.W.; Nam, E.J.; Kyung, H.S.; Park, J.Y.; Kim, I.S. LIGHT up-regulated on B lymphocytes and monocytes in rheumatoid arthritis mediates cellular adhesion and metalloproteinase production by synoviocytes. Arthritis Rheum. 2007, 56, 1106–1117. [Google Scholar] [CrossRef]
- Jubel, J.M.; Barbati, Z.R.; Burger, C.; Wirtz, D.C.; Schildberg, F.A. The role of PD-1 in acute and chronic infection. Front. Immunol. 2020, 11, 487. [Google Scholar] [CrossRef] [PubMed]
- Sharrad, D.; Chugh, P.; Slee, M.; Bacchi, S. Defining progression independent of relapse activity (PIRA) in adult patients with relapsing multiple sclerosis: A systematic review. Mult. Scler. Relat. Disord. 2023, 78, 104899. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Riddell, S.R.; Akatsuka, Y.; Jensen, M.C.; Blattman, J.N.; Greenberg, P.D. Restoration of CD28 expression in CD28- CD8+ memory effector T cells reconstitutes antigen-induced IL-2 production. J. Exp. Med. 2003, 198, 947–955. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HAMS (n = 61) | Non HAMS (n = 38) | p Value | |
|---|---|---|---|
| Demographics | |||
| Wome,. n (%) | 34 (54.8%) | 21 (56.8%) | |
| Age, years | 42.22 (10.56) | 44.41 (9.16) | 0.251 |
| Ethnicity, n (%) | 0.390 | ||
| Caucasic | 54 (88.5%) | 37 (97.4%) | |
| Hispanic | 5 (8.2%) | 1 (2.6%) | |
| African | 2 (3.3%) | - | |
| Early stage, n = 41, n (%) | 26 (63.4%) | 15 (36.6%) | |
| Long standing, n = 59, n (%) | 35 (60.3%) | 23 (39.7%) | |
| Clinical data | |||
| Baseline expanded disability status scale | 2.36 (2.27) | 1.36 (1.77) | 0.011 |
| Baseline symbol digit modality test | 41 (14.32) | 50.83 (12.07) | 0.001 |
| 3 years progression | 24 (39.9%) | 10 (27.8%) | |
| 3 years progression independent of relapse activity | 18 (75.0%) | 10 (100%) | |
| Treatments received | |||
| Interferon, n (%) | 2 (3.3%) | 5 (13.2%) | |
| Teriflunomide, n (%) | 4 (6.6%) | 2 (5.3%) | |
| S1P receptor antagonists, n (%) | 1 (1.6%) | - | |
| Dimethyl fumarate, n (%) | 5 (8.2%) | 9 (23.7%) | |
| Cladribine, n (%) | 17 (27.9%) | 14 (36.8%) | |
| Natalizumab, n (%) | 20 (32.8%) | 2 (5.3%) | |
| Anti-CD20, n (%) | 12 (19.7%) | 6 (15.8%) | |
| Previous treatments received | |||
| Interferon, n (%) | 8 (22.9%) | 6 (26.1%) | |
| Teriflunomide, n (%) | 3 (8.6%) | 3 (13.0%) | |
| Alemtuzumab, n (%) | 2 (5.7%) | - | |
| Dimethyl fumarate, n (%) | 13 (37.1%) | 4 (17.4%) | |
| Glatiramer acetate, n (%) | 2 (5.7%) | 6 (26.1%) | |
| Cladribine, n (%) | 1 (2.9%) | - | |
| Natalizumab, n (%) | 5 (14.3%) | 3 (13.0%) | |
| Anti-CD20, n (%) | 1 (2.9%) | 1 (4.3%) | |
| Interferon | Teriflunomide | Dimethyl Fumarate | Glatiramer Acetate | p Value | |
|---|---|---|---|---|---|
| B cells | 73.62 ± 12.94 | 64.62 ± 17.20 | 70.56 ± 9.63 | 68.42 ± 19.00 | 0.605 |
| HVEM effector Th | 1.00 ± 0.68 | 0.88 ± 0.61 | 1.07 ± 1.02 | 0.49 ± 0.30 | 0.409 |
| Memory CD8+ T | 16.00 ± 8.47 | 12.23 ± 8.82 | 11.34 ± 4.94 | 12.35 ± 9.35 | 0.455 |
| CD28 naïve Treg | 99.2 ± 1.47 | 99.87 ± 0.15 | 99.49 ± 0.57 | 99.77 ± 0.57 | 0.253 |
| CD28 naïve CD8+ T | 2.19 ± 1.49 | 2.19 ± 1.84 | 1.50 ± 1.59 | 2.50 ± 3.16 | 0.390 |
| HVEM classical monocytes | 0.70 ± 0.61 | 0.58 ± 0.40 | 0.7 ± 0.41 | 0.72 ±0.34 | 0.782 |
| CD28 terminal effector CD8+ | 3.90 ± 4.95 | 2.90 ± 2.78 | 3.87 ± 7.49 | 3.27 ± 5.02 | 0.509 |
| CD70 NKbright | 1.00 ± 0.89 | 0.84 ± 0.80 | 0.73 ± 0.54 | 0.41 ± 0.22 | 0.540 |
| PD-1 memory Th | 6.29 ± 2.26 | 4.82 ± 2.97 | 6.06 ± 3.54 | 5.71 ± 2.54 | 0.486 |
| HVEM plasmablasts | 6.13 ± 4.62 | 2.12 ± 0.73 | 3.27 ± 3.30 | 1.99 ± 2.44 | 0.127 |
| Variable | Initial Model | Final Model | ||
|---|---|---|---|---|
| OR (95% CI) | p Value | OR (95% CI) | p Value | |
| CD28 on terminal effector CD8+ T cells | 1.548 (0.986, 2.428) | 0.057 | 1.565 (1.081, 2.265) | 0.018 |
| CD80 on memory Treg | 1.236 (0.168, 9.100) | 0.836 | - | - |
| CTLA-4 on memory B cells | 0.570 (0.208, 1.562) | 0.275 | - | - |
| HVEM on plasmablast | 1.349 (0.943, 1.929) | 0.102 | 1.244 (0.977, 1.582) | 0.076 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
López-Molina, M.; Torres Iglesias, G.; Vidal, L.; Díaz Gamero, N.; Sánchez-Pascual, Á.; Chamorro, B.; Lozano-Rodríguez, R.; Sáenz de Santa María-Diez, G.; del Prado-Montero, J.; López-Collazo, E.; et al. Immune Checkpoint Signatures Reveal Stage-Specific Biomarkers for High-Activity Multiple Sclerosis. Int. J. Mol. Sci. 2026, 27, 1907. https://doi.org/10.3390/ijms27041907
López-Molina M, Torres Iglesias G, Vidal L, Díaz Gamero N, Sánchez-Pascual Á, Chamorro B, Lozano-Rodríguez R, Sáenz de Santa María-Diez G, del Prado-Montero J, López-Collazo E, et al. Immune Checkpoint Signatures Reveal Stage-Specific Biomarkers for High-Activity Multiple Sclerosis. International Journal of Molecular Sciences. 2026; 27(4):1907. https://doi.org/10.3390/ijms27041907
Chicago/Turabian StyleLópez-Molina, MariPaz, Gabriel Torres Iglesias, Laura Vidal, Nerea Díaz Gamero, Álvaro Sánchez-Pascual, Beatriz Chamorro, Roberto Lozano-Rodríguez, Gonzalo Sáenz de Santa María-Diez, Julia del Prado-Montero, Eduardo López-Collazo, and et al. 2026. "Immune Checkpoint Signatures Reveal Stage-Specific Biomarkers for High-Activity Multiple Sclerosis" International Journal of Molecular Sciences 27, no. 4: 1907. https://doi.org/10.3390/ijms27041907
APA StyleLópez-Molina, M., Torres Iglesias, G., Vidal, L., Díaz Gamero, N., Sánchez-Pascual, Á., Chamorro, B., Lozano-Rodríguez, R., Sáenz de Santa María-Diez, G., del Prado-Montero, J., López-Collazo, E., Díez-Tejedor, E., Laso-García, F., Gutiérrez-Fernández, M., & Otero-Ortega, L. (2026). Immune Checkpoint Signatures Reveal Stage-Specific Biomarkers for High-Activity Multiple Sclerosis. International Journal of Molecular Sciences, 27(4), 1907. https://doi.org/10.3390/ijms27041907