
Cytokine Networks in Lichen Sclerosus: A Roadmap for Diagnosis and Treatment?




Abstract

1. Introduction
2. Materials and Methods
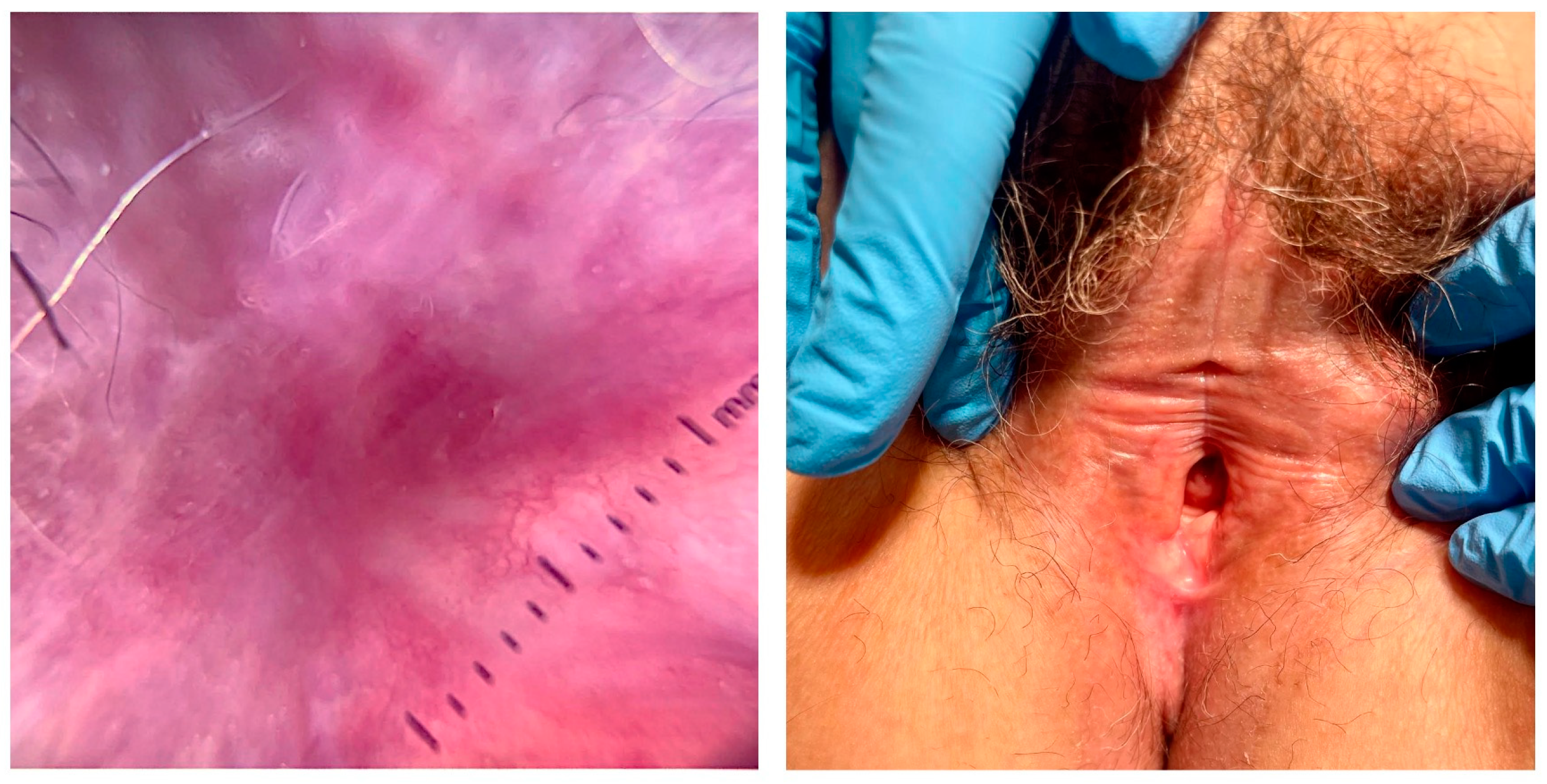
3. Diagnosis
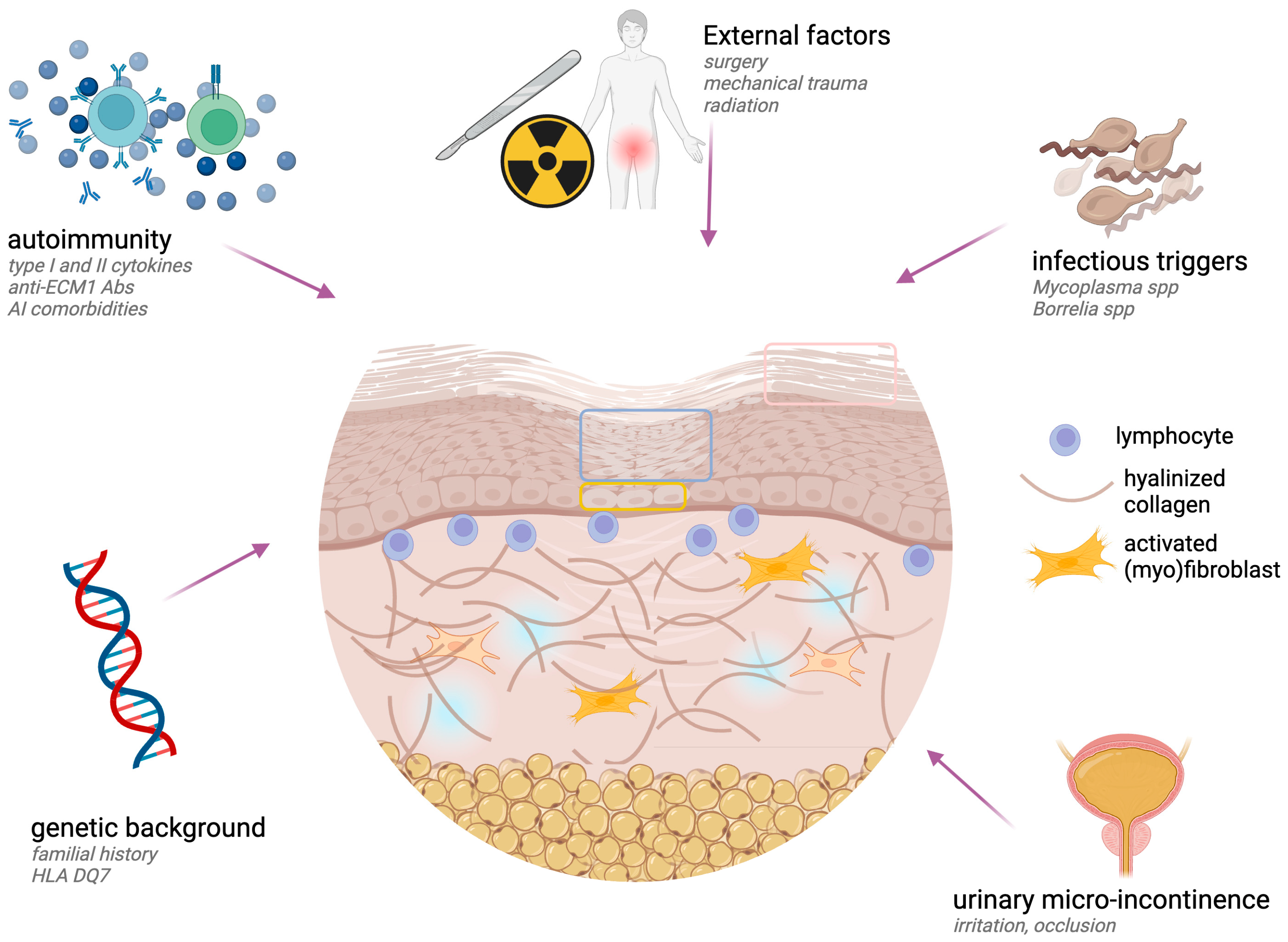
4. Etiopathogenesis
4.1. Pathogenic Events
4.2. Immune Factors and Cytokine Networks
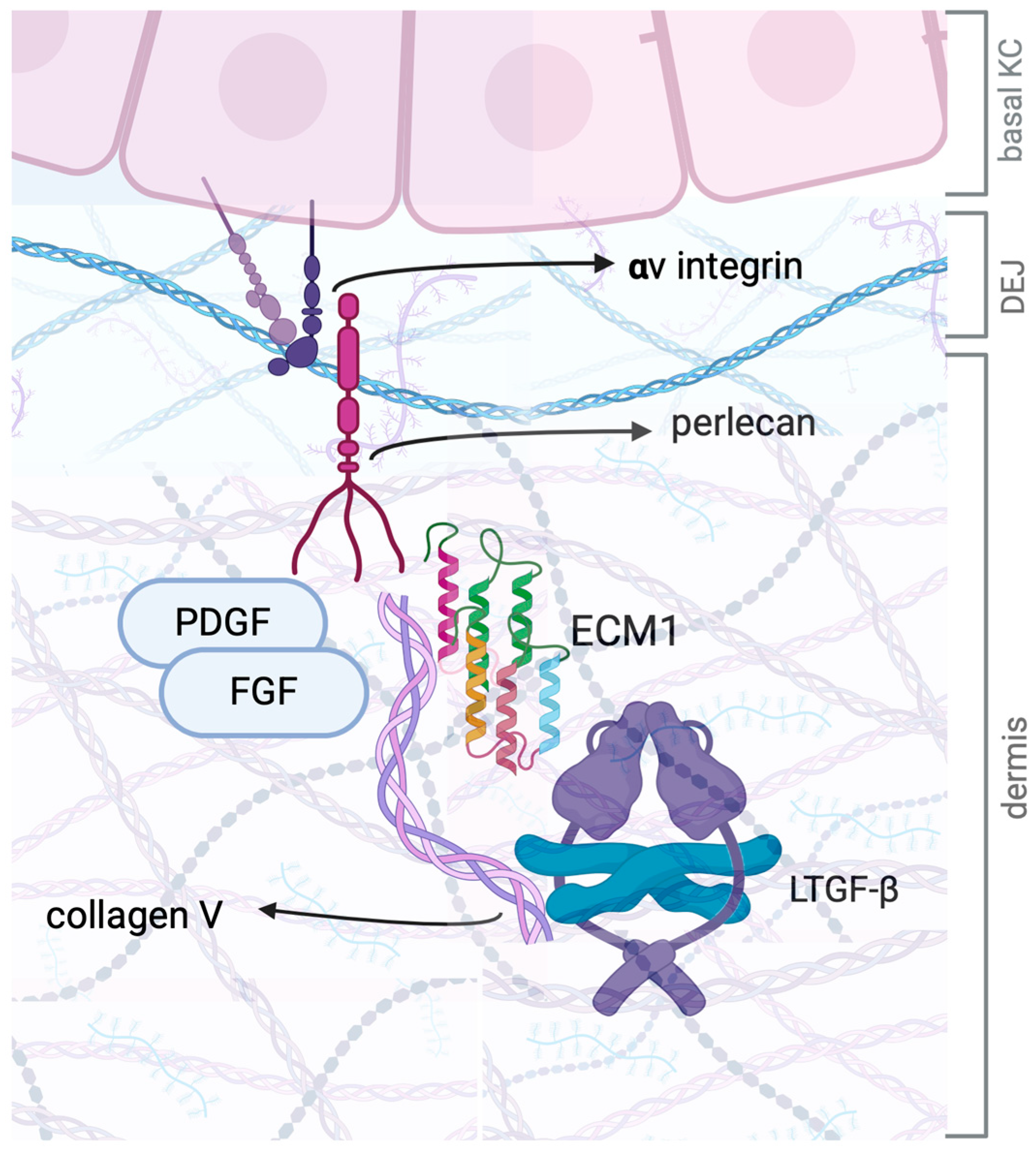
4.3. TGF-β and ECM1: A Complex Interplay in Fibrosis and Tissue Remodeling
5. Therapeutic Management: Current Guidelines and State of the Art
6. Recent Advances and Future Perspectives
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADSC | Adipose-derived stem cell |
| DEJ | Dermal–epidermal junction |
| ECM1 | Extracellular matrix protein 1 |
| FGF | Fibroblast growth factor |
| IFN-γ | Interferon gamma |
| IL | Interleukin |
| LAP | Latency-associated peptide |
| LP | Lichen planus |
| LS | Lichen sclerosus |
| NK | Natural killer cell |
| PDGF | Platelet-derived growth factor |
| PDT | Photodynamic therapy |
| PLS | Penile LS |
| PLSCR-1 | Phospholipid scramblase 1 |
| PRP | Potential of platelet-rich plasma |
| RGD | Arginine–glycine–aspartic acid |
| RT-PCR | Reverse-transcription polymerase chain reaction |
| SCC | Squamous cell carcinoma |
| TCIs | Topical calcineurin inhibitors |
| TCS | Topical corticosteroid |
| TGF-β | Transforming growth factor-beta |
| TGFBR | Transforming growth factor-beta receptor |
| Th | Helper T cells |
| TNF-α | Tumor necrosis factor alpha |
| Treg | T regulatory cell |
| VIN | Vulvar intraepithelial neoplasia |
References
- Jerkovic Gulin, S.; Liljeberg, L.; Seifert, O. The Impact of Genital Lichen Sclerosus in Men and Women on Quality of Life: A Prospective Cohort Study. Int. J. Womens Dermatol. 2024, 10, e131. [Google Scholar] [CrossRef] [PubMed]
- Kirtschig, G.; Becker, K.; Günthert, A.; Jasaitiene, D.; Cooper, S.; Chi, C.-C.; Kreuter, A.; Rall, K.K.; Aberer, W.; Riechardt, S.; et al. Evidence-Based (S3) Guideline on (Anogenital) Lichen Sclerosus. J. Eur. Acad. Dermatol. Venereol. 2015, 29, e1–e43. [Google Scholar] [CrossRef]
- Fistarol, S.K.; Itin, P.H. Diagnosis and Treatment of Lichen Sclerosus: An Update. Am. J. Clin. Dermatol. 2013, 14, 27–47. [Google Scholar] [CrossRef]
- Kirtschig, G. Lichen Sclerosus-Presentation, Diagnosis and Management. Dtsch. Arztebl. Int. 2016, 113, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Leibovitz, A.; Kaplun, V.; Saposhnicov, N.; Habot, B. Vulvovaginal Examinations in Elderly Nursing Home Women Residents. Arch. Gerontol. Geriatr. 2000, 31, 1–4. [Google Scholar] [CrossRef]
- Halonen, P.; Jakobsson, M.; Heikinheimo, O.; Gissler, M.; Pukkala, E. Incidence of Lichen Sclerosus and Subsequent Causes of Death: A Nationwide Finnish Register Study. BJOG 2020, 127, 814–819. [Google Scholar] [CrossRef]
- Kizer, W.S.; Prarie, T.; Morey, A.F. Balanitis Xerotica Obliterans: Epidemiologic Distribution in an Equal Access Health Care System. South Med. J. 2003, 96, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Niamh, L.; Naveen, S.; Hazel, B. Diagnosis of Vulval Inflammatory Dermatoses: A Pathological Study with Clinical Correlation. Int. J. Gynecol. Pathol. 2009, 28, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Bighetti, S.; Mancon, S.; Suardi, N.; Calzavara-Pinton, P.; Maione, V.; Arisi, M.; Lughezzani, G.; Zerbinati, N.; Ghini, I.; Bettolini, L. Evaluating Lichen Sclerosus in Phimosis: Insights From a Multidisciplinary Retrospective Study. Australas. J. Dermatol. 2025, 66, e39–e42. [Google Scholar] [CrossRef]
- Esse, I.; Rodriguez, K.H.; Kassels, A.; Shiu, J.; Kraus, C.N. Vulvar Lichen Sclerosus and Vitiligo: Overlap and Clinical Features. J. Am. Acad. Dermatol. 2023, 89, 839–841. [Google Scholar] [CrossRef]
- Sheinis, M.; Selk, A. Development of the Adult Vulvar Lichen Sclerosus Severity Scale—A Delphi Consensus Exercise for Item Generation. J. Low. Genit. Tract Dis. 2018, 22, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Conte, S.; Mohamed, S.D.; Cohen, Y.; Yacovelli, A.; Starkey, S.; Johnston, L.; Shergill, M.; Law, A.; Litvinov, I.V.; Mukovozov, I. Clinical Presentations and Complications of Lichen Sclerosus: A Systematic Review. J. Dtsch. Dermatol. Ges. 2025, 23, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Shasi, P.B.; Chapman, H.T.; Evans, D.T.P.; Jaleel, H. Psychological and Psychiatric Morbidity in Lichen Sclerosus in a Cohort Recruited from a Genitourinary Medicine Clinic. Int. J. STD AIDS 2010, 21, 17–18. [Google Scholar] [CrossRef]
- Ranum, A.; Pearson, D.R. The Impact of Genital Lichen Sclerosus and Lichen Planus on Quality of Life: A Review. Int. J. Womens Dermatol. 2022, 8, e042. [Google Scholar] [CrossRef]
- Moreno-Vílchez, C.; Llobera-Ris, C.; Torrecilla-Vall-Llossera, C.; Notario, J.; Figueras-Nart, I. Lichen Sclerosus and Its Association with Cancer: A Retrospective Cohort Study. Int. J. Dermatol. 2023, 62, e84–e85. [Google Scholar] [CrossRef]
- Lee, A.; Bradford, J.; Fischer, G. Long-Term Management of Adult Vulvar Lichen Sclerosus: A Prospective Cohort Study of 507 Women. JAMA Dermatol. 2015, 151, 1061–1067. [Google Scholar] [CrossRef]
- De Luca, D.A.; Papara, C.; Vorobyev, A.; Staiger, H.; Bieber, K.; Thaçi, D.; Ludwig, R.J. Lichen Sclerosus: The 2023 Update. Front. Med. 2023, 10, 1106318. [Google Scholar] [CrossRef]
- Kirtschig, G.; Kinberger, M.; Kreuter, A.; Simpson, R.; Günthert, A.; van Hees, C.; Becker, K.; Ramakers, M.J.; Corazza, M.; Müller, S.; et al. EuroGuiderm Guideline on Lichen Sclerosus-Introduction into Lichen Sclerosus. J. Eur. Acad. Dermatol. Venereol. 2024, 38, 1850–1873. [Google Scholar] [CrossRef] [PubMed]
- Lacarrubba, F.; Pellacani, G.; Verzì, A.E.; Pippione, M.; Micali, G. Extragenital Lichen Sclerosus: Clinical, Dermoscopic, Confocal Microscopy and Histologic Correlations. J. Am. Acad. Dermatol. 2015, 72, S50–S52. [Google Scholar] [CrossRef]
- Errichetti, E.; Lallas, A.; Apalla, Z.; Di Stefani, A.; Stinco, G. Dermoscopy of Morphea and Cutaneous Lichen Sclerosus: Clinicopathological Correlation Study and Comparative Analysis. Dermatology 2017, 233, 462–470. [Google Scholar] [CrossRef]
- Borghi, A.; Corazza, M.; Minghetti, S.; Bianchini, E.; Virgili, A. Dermoscopic Features of Vulvar Lichen Sclerosus in the Setting of a Prospective Cohort of Patients: New Observations. Dermatology 2016, 232, 71–77. [Google Scholar] [CrossRef]
- Huisman, B.W.; Pagan, L.; Ulrich, M.; Rissmann, R.; Damman, J.; Piek, J.M.J.; Niemeyer-van der Kolk, T.; van Poelgeest, M.I.E. Reflectance Confocal Microscopy as a Non-Invasive Imaging Tool in Vulvar High-Grade Squamous Intraepithelial Lesions and Lichen Sclerosus: A Descriptive Morphological Study in Patients and Healthy Volunteers. Exp. Dermatol. 2023, 32, 1734–1743. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Y.; Gao, X.; Qin, B.; Ren, M.; Zhang, W.; Wei, R.; Su, H.; Li, Q. In Vivo Evaluation of Vulvar Lichen Sclerosus with Reflectance Confocal Microscopy and Therapeutic Monitoring in Children. Ski. Res. Technol. 2023, 29, e13234. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.J.; Wojnarowska, F. Lichen Sclerosus. Lancet 1999, 353, 1777–1783. [Google Scholar] [CrossRef]
- Gao, X.-H.; Barnardo, M.C.M.N.; Winsey, S.; Ahmad, T.; Cook, J.; Agudelo, J.D.; Zhai, N.; Powell, J.J.; Fuggle, S.V.; Wojnarowska, F. The Association between HLA DR, DQ Antigens, and Vulval Lichen Sclerosus in the UK: HLA DRB112 and Its Associated DRB112/DQB10301/04/09/010 Haplotype Confers Susceptibility to Vulval Lichen Sclerosus, and HLA DRB10301/04 and Its Associated DRB10301/04/DQB10201/02/03 Haplotype Protects from Vulval Lichen Sclerosus. J. Investig. Dermatol. 2005, 125, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Corazza, M.; Schettini, N.; Zedde, P.; Borghi, A. Vulvar Lichen Sclerosus from Pathophysiology to Therapeutic Approaches: Evidence and Prospects. Biomedicines 2021, 9, 950. [Google Scholar] [CrossRef] [PubMed]
- Sherman, V.; McPherson, T.; Baldo, M.; Salim, A.; Gao, X.H.; Wojnarowska, F. The High Rate of Familial Lichen Sclerosus Suggests a Genetic Contribution: An Observational Cohort Study. J. Eur. Acad. Dermatol. Venereol. 2010, 24, 1031–1034. [Google Scholar] [CrossRef]
- Aslanian, F.M.N.P.; Marques, M.T.Q.; Matos, H.J.; Pontes, L.F.S.; Porto, L.C.S.; Azevedo, L.M.S.; Filgueira, A.L. HLA Markers in Familial Lichen Sclerosus. J. Dtsch. Dermatol. Ges. 2006, 4, 842–847. [Google Scholar] [CrossRef]
- Peters, D.R.; Tychsen, L. Recovery of Pupillomotor Function after Cataract Surgery. Aviat. Space Environ. Med. 1989, 60, 586–588. [Google Scholar]
- Azurdia, R.M.; Luzzi, G.A.; Byren, I.; Welsh, K.; Wojnarowska, F.; Marren, P.; Edwards, A. Lichen Sclerosus in Adult Men: A Study of HLA Associations and Susceptibility to Autoimmune Disease. Br. J. Dermatol. 1999, 140, 79–83. [Google Scholar] [CrossRef]
- Marren, P.; Yell, J.; Charnock, F.M.; Bunce, M.; Welsh, K.; Wojnarowska, F. The Association between Lichen Sclerosus and Antigens of the HLA System. Br. J. Dermatol. 1995, 132, 197–203. [Google Scholar] [CrossRef]
- Villa, M.; Dragonetti, E.; Grande, M.; Bove, P.; Sansalone, S.; Rulli, F.; Tambucci, R.; Tucci, G.; Baldi, A. Skin Phototype and Local Trauma in the Onset of Balanitis Xerotica Obliterans (BXO) in Circumcised Patients. In Vivo 2012, 26, 143–146. [Google Scholar]
- Higgins, C.A.; Cruickshank, M.E. A Population-Based Case-Control Study of Aetiological Factors Associated with Vulval Lichen Sclerosus. J. Obstet. Gynaecol. 2012, 32, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Edwards, L.R.; Privette, E.D.; Patterson, J.W.; Tchernev, G.; Chokoeva, A.A.; Wollina, U.; Lotti, T.; Wilson, B.B. Radiation-Induced Lichen Sclerosus of the Vulva: First Report in the Medical Literature. Wien. Med. Wochenschr. 2017, 167, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.-X.; Wu, X.; Chen, J.-P.; Zhou, B.-S. [Study on the risk factors of 100 cases with vulvar dystrophy]. Zhonghua Liu Xing Bing Xue Za Zhi 2003, 24, 932–934. [Google Scholar]
- Eisendle, K.; Grabner, T.; Kutzner, H.; Zelger, B. Possible Role of Borrelia Burgdorferi Sensu Lato Infection in Lichen Sclerosus. Arch. Dermatol. 2008, 144, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Pilatz, A.; Altinkilic, B.; Rusz, A.; Izykowski, N.; Traenkenschuh, W.; Rische, J.; Lehmann, U.; Herbst, C.; Maegel, L.; Becker, J.; et al. Role of Human Papillomaviruses in Persistent and Glucocorticoid-Resistant Juvenile Phimosis. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 716–721. [Google Scholar] [CrossRef]
- Nasca, M.R.; Lacarrubba, F.; Micali, G. Human Papillomavirus Infection and Lichen Sclerosus: Coincidence or Link? Int. J. Dermatol. 2018, 57, 617–618. [Google Scholar] [CrossRef]
- Mortensen, O.E.; Christensen, S.E.; Løkkegaard, E. The Evidence behind the Use of LASER for Genitourinary Syndrome of Menopause, Vulvovaginal Atrophy, Urinary Incontinence and Lichen Sclerosus: A State-of-the-Art Review. Acta Obstet. Gynecol. Scand. 2022, 101, 657–692. [Google Scholar] [CrossRef]
- Kirby, L.; Gran, S.; Kreuser-Genis, I.; Owen, C.; Simpson, R. Is Urinary Incontinence Associated with Lichen Sclerosus in Females? A Systematic Review and Meta-Analysis. Ski. Health Dis. 2021, 1, ski2.13. [Google Scholar] [CrossRef]
- Halonen, P.; Heikinheimo, O.; Hadkhale, K.; Gissler, M.; Pukkala, E.; Jakobsson, M. Risk Factors for Lichen Sclerosus: A Case-Control Study of 43,000 Finnish Women. J. Low. Genit. Tract Dis. 2024, 28, 164–168. [Google Scholar] [CrossRef]
- Panou, E.; Panagou, E.; Foley, C.; Kravvas, G.; Watchorn, R.; Alnajjar, H.; Muneer, A.; Bunker, C.B. Male Genital Lichen Sclerosus Associated with Urological Interventions and Microincontinence: A Case Series of 21 Patients. Clin. Exp. Dermatol. 2022, 47, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Kravvas, G.; Muneer, A.; Watchorn, R.E.; Castiglione, F.; Haider, A.; Freeman, A.; Hadway, P.; Alnajjar, H.; Lynch, M.; Bunker, C.B. Male Genital Lichen Sclerosus, Microincontinence and Occlusion: Mapping the Disease across the Prepuce. Clin. Exp. Dermatol. 2022, 47, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Malhotra, A.K.; Ajith, C. Lichen Sclerosus: Role of Occlusion of the Genital Skin in the Pathogenesis. Indian J. Dermatol. Venereol. Leprol. 2010, 76, 56–58. [Google Scholar] [CrossRef]
- Gambichler, T.; Belz, D.; Terras, S.; Kreuter, A. Humoral and Cell-Mediated Autoimmunity in Lichen Sclerosus. Br. J. Dermatol. 2013, 169, 183–184. [Google Scholar] [CrossRef]
- Farrell, A.M.; Dean, D.; Millard, P.R.; Charnock, F.M.; Wojnarowska, F. Cytokine Alterations in Lichen Sclerosus: An Immunohistochemical Study. Br. J. Dermatol. 2006, 155, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Tchórzewski, H.; Rotsztejn, H.; Banasik, M.; Lewkowicz, P.; Głowacka, E. The Involvement of Immunoregulatory T Cells in the Pathogenesis of Lichen Sclerosus. Med. Sci. Monit. 2005, 11, CR39–CR43. [Google Scholar]
- Czajkowski, M.; Wierzbicki, P.; Kotulak-Chrząszcz, A.; Czajkowska, K.; Bolcewicz, M.; Kłącz, J.; Kreft, K.; Lewandowska, A.; Nedoszytko, B.; Sokołowska-Wojdyło, M.; et al. The Role of Occlusion and Micro-Incontinence in the Pathogenesis of Penile Lichen Sclerosus: An Observational Study of pro-Inflammatory Cytokines’ Gene Expression. Int. Urol. Nephrol. 2022, 54, 763–772. [Google Scholar] [CrossRef]
- Courtney, A.; Adamson, S.R.; Veysey, E. Adalimumab Use in Severe Recalcitrant Vulval Lichen Sclerosus and Vulval Lichen Planus. J. Low. Genit. Tract Dis. 2025, 29, 190–194. [Google Scholar] [CrossRef]
- Clay, F.E.; Cork, M.J.; Tarlow, J.K.; Blakemore, A.I.; Harrington, C.I.; Lewis, F.; Duff, G.W. Interleukin 1 Receptor Antagonist Gene Polymorphism Association with Lichen Sclerosus. Hum. Genet. 1994, 94, 407–410. [Google Scholar] [CrossRef]
- Terlou, A.; Santegoets, L.A.M.; van der Meijden, W.I.; Heijmans-Antonissen, C.; Swagemakers, S.M.A.; van der Spek, P.J.; Ewing, P.C.; van Beurden, M.; Helmerhorst, T.J.M.; Blok, L.J. An Autoimmune Phenotype in Vulvar Lichen Sclerosus and Lichen Planus: A Th1 Response and High Levels of microRNA-155. J. Investig. Dermatol. 2012, 132, 658–666. [Google Scholar] [CrossRef]
- Peterson, D.M.; Damsky, W.E.; Vesely, M.D. Treatment of Lichen Sclerosus and Hypertrophic Scars with Dupilumab. JAAD Case Rep. 2022, 23, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Carli, P.; Moretti, S.; Spallanzani, A.; Berti, E.; Cattaneo, A. Fibrogenic Cytokines in Vulvar Lichen Sclerosus. An Immunohistochemical Study. J. Reprod. Med. 1997, 42, 161–165. [Google Scholar]
- Baran, W.; Woźniak, Z.; Batycka-Baran, A. IL-17: A Novel Player in the Pathogenesis of Vulvar Lichen Sclerosus. Postep. Dermatol Alergol 2024, 41, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Moreau, J.M.; Velegraki, M.; Bolyard, C.; Rosenblum, M.D.; Li, Z. Transforming Growth Factor-Β1 in Regulatory T Cell Biology. Sci. Immunol. 2022, 7, eabi4613. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Cheng, G. Role of Interleukin 10 Transcriptional Regulation in Inflammation and Autoimmune Disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef]
- Soltanzadeh-Yamchi, M.; Shahbazi, M.; Aslani, S.; Mohammadnia-Afrouzi, M. MicroRNA Signature of Regulatory T Cells in Health and Autoimmunity. Biomed. Pharmacother. 2018, 100, 316–323. [Google Scholar] [CrossRef]
- Divekar, A.A.; Dubey, S.; Gangalum, P.R.; Singh, R.R. Dicer Insufficiency and microRNA-155 Overexpression in Lupus Regulatory T Cells: An Apparent Paradox in the Setting of an Inflammatory Milieu. J. Immunol. 2011, 186, 924–930. [Google Scholar] [CrossRef]
- Stahl, H.F.; Fauti, T.; Ullrich, N.; Bopp, T.; Kubach, J.; Rust, W.; Labhart, P.; Alexiadis, V.; Becker, C.; Hafner, M.; et al. miR-155 Inhibition Sensitizes CD4+ Th Cells for TREG Mediated Suppression. PLoS ONE 2009, 4, e7158. [Google Scholar] [CrossRef]
- Corazza, M.; Oton-Gonzalez, L.; Scuderi, V.; Rotondo, J.C.; Lanzillotti, C.; Di Mauro, G.; Tognon, M.; Martini, F.; Borghi, A. Tissue Cytokine/Chemokine Profile in Vulvar Lichen Sclerosus: An Observational Study on Keratinocyte and Fibroblast Cultures. J. Dermatol. Sci. 2020, 100, 223–226. [Google Scholar] [CrossRef]
- Wang, H.; Wu, J.; Ma, L.; Bai, Y.; Liu, J. The Role of Interleukin -1 Family in Fibrotic Diseases. Cytokine 2023, 165, 156161. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hur, H.; Ashkenazi, M.; Huszar, M.; Gurevich, P.; Zusman, I. Lymphoid Elements and Apoptosis-Related Proteins (Fas, Fas Ligand, P53 and Bcl-2) in Lichen Sclerosus and Carcinoma of the Vulva. Eur. J. Gynaecol. Oncol. 2001, 22, 104–109. [Google Scholar]
- Tran, D.A.; Tan, X.; Macri, C.J.; Goldstein, A.T.; Fu, S.W. Lichen Sclerosus: An Autoimmunopathogenic and Genomic Enigma with Emerging Genetic and Immune Targets. Int. J. Biol. Sci. 2019, 15, 1429–1439. [Google Scholar] [CrossRef]
- Dufour, A.M.; Borowczyk-Michalowska, J.; Alvarez, M.; Truchetet, M.-E.; Modarressi, A.; Brembilla, N.C.; Chizzolini, C. IL-17A Dissociates Inflammation from Fibrogenesis in Systemic Sclerosis. J. Investig. Dermatol. 2020, 140, 103–112.E8. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Abraham, D.; Ong, V. The Yin and Yang of IL-17 in Systemic Sclerosis. Front. Immunol. 2022, 13, 885609. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.J.; Sullivan, B.M.; Peng, S.L.; Glimcher, L.H. Molecular Mechanisms Regulating Th1 Immune Responses. Annu. Rev. Immunol. 2003, 21, 713–758. [Google Scholar] [CrossRef]
- Valencia, X.; Stephens, G.; Goldbach-Mansky, R.; Wilson, M.; Shevach, E.M.; Lipsky, P.E. TNF Downmodulates the Function of Human CD4+CD25hi T-Regulatory Cells. Blood 2006, 108, 253–261. [Google Scholar] [CrossRef]
- Romero, L.I.; Pincus, S.H. In Situ Localization of Interleukin-6 in Normal Skin and Atrophic Cutaneous Disease. Int. Arch. Allergy Immunol. 1992, 99, 44–49. [Google Scholar] [CrossRef]
- Johnson, B.Z.; Stevenson, A.W.; Prêle, C.M.; Fear, M.W.; Wood, F.M. The Role of IL-6 in Skin Fibrosis and Cutaneous Wound Healing. Biomedicines 2020, 8, 101. [Google Scholar] [CrossRef]
- Corthay, A. How Do Regulatory T Cells Work? Scand. J. Immunol. 2009, 70, 326–336. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T Cells and Immune Tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, N.; Terlizzi, J.; Aho, S.; Brittingham, R.; Fertala, A.; Oyama, N.; McGrath, J.A.; Uitto, J. Extracellular Matrix Protein 1 Inhibits the Activity of Matrix Metalloproteinase 9 through High-Affinity Protein/Protein Interactions. Exp. Dermatol. 2006, 15, 300–307. [Google Scholar] [CrossRef]
- Bhalerao, J.; Tylzanowski, P.; Filie, J.D.; Kozak, C.A.; Merregaert, J. Molecular Cloning, Characterization, and Genetic Mapping of the cDNA Coding for a Novel Secretory Protein of Mouse. Demonstration of Alternative Splicing in Skin and Cartilage. J. Biol. Chem. 1995, 270, 16385–16394. [Google Scholar] [CrossRef]
- Chan, I. The Role of Extracellular Matrix Protein 1 in Human Skin. Clin. Exp. Dermatol. 2004, 29, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Hamada, T.; McLean, W.H.I.; Ramsay, M.; Ashton, G.H.S.; Nanda, A.; Jenkins, T.; Edelstein, I.; South, A.P.; Bleck, O.; Wessagowit, V.; et al. Lipoid Proteinosis Maps to 1q21 and Is Caused by Mutations in the Extracellular Matrix Protein 1 Gene (ECM1). Hum. Mol. Genet. 2002, 11, 833–840. [Google Scholar] [CrossRef]
- Hamada, T. Lipoid Proteinosis. Clin. Exp. Dermatol. 2002, 27, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Oyama, N.; Chan, I.; Neill, S.M.; Hamada, T.; South, A.P.; Wessagowit, V.; Wojnarowska, F.; D’Cruz, D.; Hughes, G.J.; Black, M.M.; et al. Autoantibodies to Extracellular Matrix Protein 1 in Lichen Sclerosus. Lancet 2003, 362, 118–123. [Google Scholar] [CrossRef]
- Edmonds, E.V.J.; Oyama, N.; Chan, I.; Francis, N.; McGrath, J.A.; Bunker, C.B. Extracellular Matrix Protein 1 Autoantibodies in Male Genital Lichen Sclerosus: Correspondence. Br. J. Dermatol. 2011, 165, 218–219. [Google Scholar] [CrossRef]
- Mongiat, M.; Fu, J.; Oldershaw, R.; Greenhalgh, R.; Gown, A.M.; Iozzo, R.V. Perlecan Protein Core Interacts with Extracellular Matrix Protein 1 (ECM1), a Glycoprotein Involved in Bone Formation and Angiogenesis. J. Biol. Chem. 2003, 278, 17491–17499. [Google Scholar] [CrossRef]
- Merregaert, J.; Van Langen, J.; Hansen, U.; Ponsaerts, P.; El Ghalbzouri, A.; Steenackers, E.; Van Ostade, X.; Sercu, S. Phospholipid Scramblase 1 Is Secreted by a Lipid Raft-Dependent Pathway and Interacts with the Extracellular Matrix Protein 1 in the Dermal Epidermal Junction Zone of Human Skin. J. Biol. Chem. 2010, 285, 37823–37837. [Google Scholar] [CrossRef]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. Growth Factors and Cytokines in Wound Healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Utsunomiya, N.; Utsunomiya, A.; Chino, T.; Hasegawa, M.; Oyama, N. Gene Silencing of Extracellular Matrix Protein 1 (ECM1) Results in Phenotypic Alterations of Dermal Fibroblasts Reminiscent of Clinical Features of Lichen Sclerosus. J. Dermatol. Sci. 2020, 100, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Godoy, C.A.P.; Teodoro, W.R.; Velosa, A.P.P.; Garippo, A.L.; Eher, E.M.; Parra, E.R.; Sotto, M.N.; Capelozzi, V.L. Unusual Remodeling of the Hyalinization Band in Vulval Lichen Sclerosus by Type V Collagen and ECM 1 Protein. Clinics 2015, 70, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Symoens, S.; Renard, M.; Bonod-Bidaud, C.; Syx, D.; Vaganay, E.; Malfait, F.; Ricard-Blum, S.; Kessler, E.; Van Laer, L.; Coucke, P.; et al. Identification of Binding Partners Interacting with the A1-N-Propeptide of Type V Collagen. Biochem. J. 2011, 433, 371–381. [Google Scholar] [CrossRef]
- Mak, K.M.; Png, C.Y.M.; Lee, D.J. Type V Collagen in Health, Disease, and Fibrosis. Anat. Rec. 2016, 299, 613–629. [Google Scholar] [CrossRef]
- Fan, W.; Liu, T.; Chen, W.; Hammad, S.; Longerich, T.; Hausser, I.; Fu, Y.; Li, N.; He, Y.; Liu, C.; et al. ECM1 Prevents Activation of Transforming Growth Factor β, Hepatic Stellate Cells, and Fibrogenesis in Mice. Gastroenterology 2019, 157, 1352–1367.e13. [Google Scholar] [CrossRef]
- Yoshinaga, K.; Obata, H.; Jurukovski, V.; Mazzieri, R.; Chen, Y.; Zilberberg, L.; Huso, D.; Melamed, J.; Prijatelj, P.; Todorovic, V.; et al. Perturbation of Transforming Growth Factor (TGF)-Beta1 Association with Latent TGF-Beta Binding Protein Yields Inflammation and Tumors. Proc. Natl. Acad. Sci. USA 2008, 105, 18758–18763. [Google Scholar] [CrossRef]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The Master Regulator of Fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Pilatz, A.; Altinkilic, B.; Schormann, E.; Maegel, L.; Izykowski, N.; Becker, J.; Weidner, W.; Kreipe, H.; Jonigk, D. Congenital Phimosis in Patients with and without Lichen Sclerosus: Distinct Expression Patterns of Tissue Remodeling Associated Genes. J. Urol. 2013, 189, 268–274. [Google Scholar] [CrossRef]
- Sun, P.; Kraus, C.N.; Zhao, W.; Xu, J.; Suh, S.; Nguyen, Q.; Jia, Y.; Nair, A.; Oakes, M.; Tinoco, R.; et al. Single-Cell and Spatial Transcriptomics of Vulvar Lichen Sclerosus Reveal Multi-Compartmental Alterations in Gene Expression and Signaling Cross-Talk. bioRxiv 2024. [Google Scholar] [CrossRef]
- Link, F.; Li, Y.; Zhao, J.; Munker, S.; Fan, W.; Nwosu, Z.C.; Yao, Y.; Wang, S.; Huang, C.; Liebe, R.; et al. ECM1 Attenuates Hepatic Fibrosis by Interfering with Mediators of Latent TGF-Β1 Activation. Gut 2025, 74, 424–439. [Google Scholar] [CrossRef]
- Akel, R.; Fuller, C. Updates in Lichen Sclerosis: British Association of Dermatologists Guidelines for the Management of Lichen Sclerosus 2018. Br. J. Dermatol. 2018, 178, 823–824. [Google Scholar] [CrossRef]
- Kirtschig, G.; Kinberger, M.; Kreuter, A.; Simpson, R.; Günthert, A.; van Hees, C.; Becker, K.; Ramakers, M.J.; Corazza, M.; Müller, S.; et al. EuroGuiderm Guideline on Lichen Sclerosus-Treatment of Lichen Sclerosus. J. Eur. Acad. Dermatol. Venereol. 2024, 38, 1874–1909. [Google Scholar] [CrossRef] [PubMed]
- Virgili, A.; Minghetti, S.; Borghi, A.; Corazza, M. Proactive Maintenance Therapy with a Topical Corticosteroid for Vulvar Lichen Sclerosus: Preliminary Results of a Randomized Study. Br. J. Dermatol. 2013, 168, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Yesudian, P.D. The Role of Calcineurin Inhibitors in the Management of Lichen Sclerosus. Am. J. Clin. Dermatol. 2009, 10, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Virgili, A.; Minghetti, S.; Borghi, A.; Corazza, M. Long-Term Maintenance Therapy for Vulvar Lichen Sclerosus: The Results of a Randomized Study Comparing Topical Vitamin E with an Emollient. Eur. J. Dermatol. 2013, 23, 189–194. [Google Scholar] [CrossRef]
- Chi, C.-C.; Kirtschig, G.; Baldo, M.; Brackenbury, F.; Lewis, F.; Wojnarowska, F. Topical Interventions for Genital Lichen Sclerosus. Cochrane Database Syst. Rev. 2011, 2011, CD008240. [Google Scholar] [CrossRef]
- Rijal, H.; Sood, S.; Bagit, A.; Maliyar, K.; Georgakopoulos, J.R.; Mufti, A.; Yeung, J. Biologics and Small Molecules in Lichen Sclerosus: A Systematic Review. J. Cutan. Med. Surg. 2025, 12034754251316490. [Google Scholar] [CrossRef]
- Knobler, R.; Moinzadeh, P.; Hunzelmann, N.; Kreuter, A.; Cozzio, A.; Mouthon, L.; Cutolo, M.; Rongioletti, F.; Denton, C.P.; Rudnicka, L.; et al. European Dermatology Forum S1-Guideline on the Diagnosis and Treatment of Sclerosing Diseases of the Skin, Part 1: Localized Scleroderma, Systemic Sclerosis and Overlap Syndromes. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1401–1424. [Google Scholar] [CrossRef]
- Gerkowicz, A.; Szczepanik-Kułak, P.; Krasowska, D. Photodynamic Therapy in the Treatment of Vulvar Lichen Sclerosus: A Systematic Review of the Literature. J. Clin. Med. 2021, 10, 5491. [Google Scholar] [CrossRef]
- Prodromidou, A.; Chatziioannou, E.; Daskalakis, G.; Stergios, K.; Pergialiotis, V. Photodynamic Therapy for Vulvar Lichen Sclerosus-A Systematic Review. J. Low. Genit. Tract Dis. 2018, 22, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Paganelli, A.; Contu, L.; Condorelli, A.; Ficarelli, E.; Motolese, A.; Paganelli, R.; Motolese, A. Platelet-Rich Plasma (PRP) and Adipose-Derived Stem Cell (ADSC) Therapy in the Treatment of Genital Lichen Sclerosus: A Comprehensive Review. Int. J. Mol. Sci. 2023, 24, 16107. [Google Scholar] [CrossRef] [PubMed]
- Paganelli, A.; Contu, L.; Ficarelli, E.; Garbarino, F.; Motolese, A. Management of Lichen Sclerosus and Related Comorbidities at a Tertiary Referral Center: Beyond Topical Steroids. Dermatol. Pract. Concept. 2024, 14, e2024262. [Google Scholar] [CrossRef]
- Paganelli, A.; Contu, L.; Ficarelli, E.; Garbarino, F.; Motolese, A. Platelet-Rich Plasma and Stromal Vascular Fraction as Regenerative Strategies for Lichen Sclerosus: A Single-Center Experience. Int. J. Dermatol. 2024, 63, e452–e454. [Google Scholar] [CrossRef] [PubMed]
- Paganelli, A.; Fabbri, P.V.; Ghidini, F.; Bigi, L.; Lasagni, C.; Ceccarelli, P.L. Treatment and Follow-up of Genital Lichen Sclerosus in Male Children: Multidisciplinary Management at a Tertiary Care Center. Dermatol. Rep. 2023, 15, 9774. [Google Scholar] [CrossRef]
- Andugulapati, S.B.; Gourishetti, K.; Tirunavalli, S.K.; Shaikh, T.B.; Sistla, R. Biochanin-A Ameliorates Pulmonary Fibrosis by Suppressing the TGF-β Mediated EMT, Myofibroblasts Differentiation and Collagen Deposition in in Vitro and in Vivo Systems. Phytomedicine 2020, 78, 153298. [Google Scholar] [CrossRef]
- Ong, C.H.; Tham, C.L.; Harith, H.H.; Firdaus, N.; Israf, D.A. TGF-β-Induced Fibrosis: A Review on the Underlying Mechanism and Potential Therapeutic Strategies. Eur. J. Pharmacol. 2021, 911, 174510. [Google Scholar] [CrossRef]
- Niu, X.; Chang, W.; Liu, R.; Hou, R.; Li, J.; Wang, C.; Li, X.; Zhang, K. mRNA and Protein Expression of the Angiogenesis-Related Genes EDIL3, AMOT and ECM1 in Mesenchymal Stem Cells in Psoriatic Dermis. Clin. Exp. Dermatol. 2016, 41, 533–540. [Google Scholar] [CrossRef]
- Zundell, M.P.; Al-Dehneem, R.; Song, T.; Yousif, J.; Gottlieb, A.B. Novel Clinical Applications of Topical Ruxolitinib: A Case Series. J. Drugs Dermatol. 2024, 23, 188–190. [Google Scholar] [CrossRef]
- Liu, L.; Zhan, Y.; Shi, Y.; Zeng, Z.; Yu, J.; Zou, P.; Qiu, X.; Zhou, Y.; Zhang, G.; Ding, Y.; et al. Bullous Lichen Sclerosus-Generalized Morphea Overlap Syndrome Improved by Tofacitinib. Dermatol. Ther. 2022, 35, e15942. [Google Scholar] [CrossRef]
- Kirk, P.S.; Yi, Y.; Hadj-Moussa, M.; Malaeb, B.S. Diversity of Patient Profile, Urethral Stricture, and Other Disease Manifestations in a Cohort of Adult Men with Lichen Sclerosus. Investig. Clin. Urol. 2016, 57, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Kim, E.H.; Lee, M.; Jung, I.; Ahn, S.S. Risk of Cancer, Tuberculosis and Serious Infections in Patients with Ankylosing Spondylitis, Psoriatic Arthritis and Psoriasis Treated with IL-17 and TNF-α Inhibitors: A Nationwide Nested Case-Control Analysis. Clin. Exp. Rheumatol. 2023, 41, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Kim, S.R.; Kim, Y.-J.; Park, S.J.; Jeong, S.; Park, S.M. Risk of Hematologic Malignancies in Psoriasis and Rheumatoid Arthritis Patients Using Long Term TNF-α Inhibitors: A Retrospective Nationwide Study. Sci. Rep. 2025, 15, 7949. [Google Scholar] [CrossRef]
- Mansilla-Polo, M.; Morgado-Carrasco, D. Biologics Versus JAK Inhibitors. Part I: Cancer Risk. A Narrative Review. Dermatol. Ther. 2024, 14, 1389–1442. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-H.; Shen, D.; Chan, T.C.; Cho, Y.-T.; Tang, C.-H.; Chu, C.-Y. Association Between the Use of Topical Calcineurin Inhibitors and the Risk of Cancer Among Patients with Atopic Dermatitis: A Nationwide, Population-Based, Retrospective Cohort Study. Am. J. Clin. Dermatol. 2023, 24, 799–808. [Google Scholar] [CrossRef]
- Nakamura, T.; Jinta, T.; Kitamura, A.; Tanaka, M.; So, C.; Ro, S.; Imai, R.; Okafuji, K.; Tomishima, Y.; Nishimura, N. Differences in Tolerability of Antifibrotic Agents Between Connective Tissue Disease-Associated and Non-Connective Tissue Disease-Associated Interstitial Lung Disease. Cureus 2025, 17, e78750. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokine | Expression | Main Cell Source | Significance | References |
|---|---|---|---|---|
| IL-1α/β | Increased | Th1 and Th17 cells, macrophages, keratinocytes | Drives inflammation in both innate and adaptive immunity; contributes to fibrosis | Carli et al. [53] Farrell et al. [46] Czajkowski et al. [48] Wang et al. [61] |
| IL-2 | Increased | Th1 cells | Promotes T cell proliferation and immune activation | Ben-Hur et al. [62] Tchórzewski et al. [47] Farrell et al. [46] |
| IL-7 | Increased | Stromal and dendritic cells | Essential for T cell development and survival | Tran et al. [63] De Luca et al. [17] |
| IL-15 | Increased | Dendritic cells, macrophages, epithelial cells | Stimulates NK cells and memory CD8+ T cell survival and proliferation | Tran et al. [63] De Luca et al. [17] |
| IL-17 | Increased | Th17 cells, NK cells, γδ T cells | Promotes autoimmunity, activates keratinocytes, and has a controversial role in fibrosis | Dufour et al. [64] Wei et al. [65] Baran et al. [54] |
| IFN-γ | Increased | Th1 and NK cells | Activates macrophages and other immune cells, sustaining inflammation in LS | Carli et al. [53] Farrell et al. [46] Czajkowski et al. [48] |
| TNF-α | Increased | Th1 and NK cells, stressed skin cells | Regulates Treg function and immune cell recruitment, driving inflammation | Szabo et al. [66] Farrell et al. [46] Valencia et al. [67] Terlou et al. [51] Gambichler et al. [45] |
| IL-6 | Increased | Th1 cells | Drives inflammation, modulates Treg function, and promotes fibrosis through TGF-β | Romero et al. [68] Farrell et al. [46] Gambichler et al. [45] Johnson et al. [69] Czajkowski et al. [48] |
| IL-4 | Increased * | Th2 cells | Mediates pruritus and fibrosis through Th2 responses and collagen production | Carli et al. [53] Terlou et al. [51] Peterson et al. [52] |
| IL-10 | Decreased | Treg cells | Downstream marker of Treg function, suppressed by the overexpression of miR-155 | Corthay [70] Sakaguchi et al. [71] Gambichler et al. [45] |
| TGF-β | Equal | Th1 cells, Th17 cells, fibroblasts | Key mediator of fibrosis and Treg cell function in skin | Carli et al. [53] Fujimoto et al. [72] Gambichler et al. [45] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paganelli, A.; Didona, D.; Scala, E. Cytokine Networks in Lichen Sclerosus: A Roadmap for Diagnosis and Treatment? Int. J. Mol. Sci. 2025, 26, 4315. https://doi.org/10.3390/ijms26094315
Paganelli A, Didona D, Scala E. Cytokine Networks in Lichen Sclerosus: A Roadmap for Diagnosis and Treatment? International Journal of Molecular Sciences. 2025; 26(9):4315. https://doi.org/10.3390/ijms26094315
Chicago/Turabian StylePaganelli, Alessia, Dario Didona, and Emanuele Scala. 2025. "Cytokine Networks in Lichen Sclerosus: A Roadmap for Diagnosis and Treatment?" International Journal of Molecular Sciences 26, no. 9: 4315. https://doi.org/10.3390/ijms26094315
APA StylePaganelli, A., Didona, D., & Scala, E. (2025). Cytokine Networks in Lichen Sclerosus: A Roadmap for Diagnosis and Treatment? International Journal of Molecular Sciences, 26(9), 4315. https://doi.org/10.3390/ijms26094315