
Innovations in Drug Discovery for Sickle Cell Disease Targeting Oxidative Stress and NRF2 Activation—A Short Review


Abstract
:1. Introduction
2. Oxidative Stress
3. Electron Transport Chain
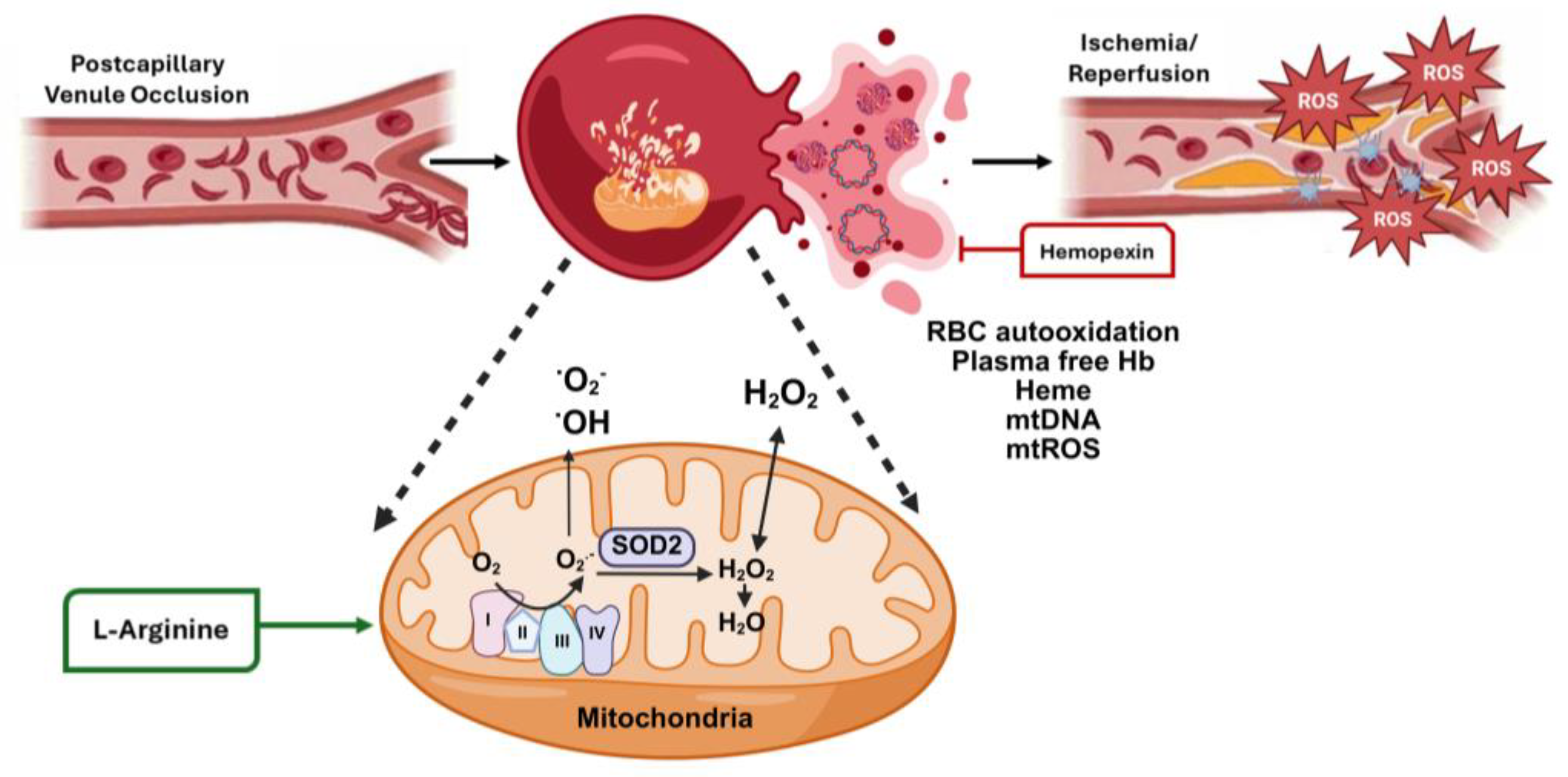
4. Oxidative Stress in Sickle Cell Disease
Environmental Factors
5. Murine Studies Linking Oxidative Stress to SCD Clinical Severity
6. Targeted Therapy to Reduce Oxidative Stress in SCD
6.1. N-Acetyl Cysteine (NAC)
6.2. Hemopexin
6.3. Alpha-Lipoic Acid
6.4. δ-Aminolevulinate
6.5. L-Arginine
6.6. Sulforaphane
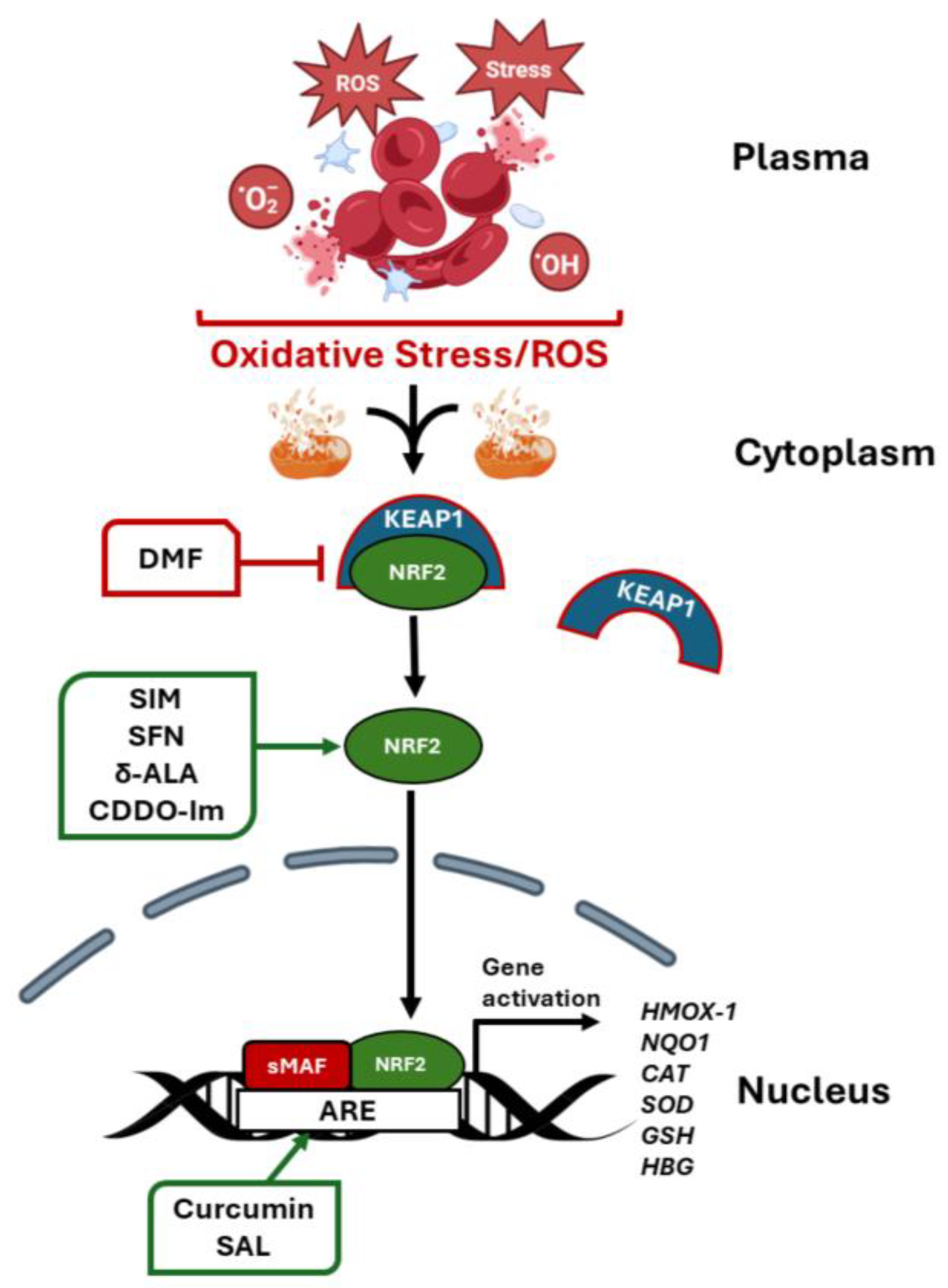
7. Role of NRF2 in Globin Gene Regulation
7.1. BACH 1 Inhibitors
7.2. Regulation by MicroRNA Genes
8. Future Directions
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Royal, C.D.M.; Babyak, M.; Shah, N.; Srivatsa, S.; Stewart, K.A.; Tanabe, P.; Wonkam, A.; Asnani, M. Sickle cell disease is a global prototype for integrative research and healthcare. Adv. Genet. 2021, 2, e10037. [Google Scholar] [CrossRef] [PubMed]
- Vona, R.; Sposi, N.M.; Mattia, L.; Gambardella, L.; Straface, E.; Pietraforte, D. Sickle Cell Disease: Role of Oxidative Stress and Antioxidant Therapy. Antioxidants 2021, 10, 296. [Google Scholar] [CrossRef] [PubMed]
- Piel, F.B.; Rees, D.C.; DeBaun, M.R.; Nnodu, O.; Ranque, B.; Thompson, A.A.; Ware, R.E.; Abboud, M.R.; Abraham, A.; Ambrose, E.E.; et al. Defining global strategies to improve outcomes in sickle cell disease: A Lancet Haematology Commission. Lancet Haematol. 2023, 10, e633–e686. [Google Scholar] [CrossRef]
- Hoban, M.D.; Orkin, S.H.; Bauer, D.E. Genetic treatment of a molecular disorder: Gene therapy approaches to sickle cell disease. Blood 2016, 127, 839–848. [Google Scholar] [CrossRef]
- Stamatoyannopoulos, G. Human hemoglobin switching. Science 1991, 252, 383. [Google Scholar] [CrossRef]
- Ferrone, F.A.; Hofrichter, J.; Eaton, W.A. Kinetics of sickle hemoglobin polymerization. I. Studies using temperature-jump and laser photolysis techniques. J. Mol. Biol. 1985, 183, 591–610. [Google Scholar] [CrossRef]
- Stamatoyannopoulos, G. The molecular basis of hemoglobin disease. Annu. Rev. Genet. 1972, 6, 47–70. [Google Scholar] [CrossRef]
- Sharpe, C.C.; Thein, S.L. Sickle cell nephropathy—A practical approach. Br. J. Haematol. 2011, 155, 287–297. [Google Scholar] [CrossRef]
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Investig. 2017, 127, 750–760. [Google Scholar] [CrossRef]
- Potoka, K.P.; Gladwin, M.T. Vasculopathy and pulmonary hypertension in sickle cell disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L314–L324. [Google Scholar] [CrossRef]
- Nur, E.; Biemond, B.J.; Otten, H.M.; Brandjes, D.P.; Schnog, J.J.; Group, C.S. Oxidative stress in sickle cell disease; pathophysiology and potential implications for disease management. Am. J. Hematol. 2011, 86, 484–489. [Google Scholar] [CrossRef]
- Tumburu, L.; Ghosh-Choudhary, S.; Seifuddin, F.T.; Barbu, E.A.; Yang, S.; Ahmad, M.M.; Wilkins, L.H.W.; Tunc, I.; Sivakumar, I.; Nichols, J.S.; et al. Circulating mitochondrial DNA is a proinflammatory DAMP in sickle cell disease. Blood 2021, 137, 3116–3126. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswaran, R.; Vazquez, B.A.; Thiruppathi, M.; Ganesh, B.B.; Ibanez, V.; Cui, S.; Engel, J.D.; Diamond, A.M.; Molokie, R.E.; DeSimone, J.; et al. Pharmacological inhibition of LSD1 and mTOR reduces mitochondrial retention and associated ROS levels in the red blood cells of sickle cell disease. Exp. Hematol. 2017, 50, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Gallivan, A.; Alejandro, M.; Kanu, A.; Zekaryas, N.; Horneman, H.; Hong, L.K.; Vinchinsky, E.; Lavelle, D.; Diamond, A.M.; Molokie, R.E.; et al. Reticulocyte mitochondrial retention increases reactive oxygen species and oxygen consumption in mouse models of sickle cell disease and phlebotomy-induced anemia. Exp. Hematol. 2023, 122, 55–62. [Google Scholar] [CrossRef]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Aspects Med. 2011, 32, 234–246. [Google Scholar] [CrossRef]
- Suzuki, T.; Takahashi, J.; Yamamoto, M. Molecular Basis of the KEAP1-NRF2 Signaling Pathway. Mol. Cells 2023, 46, 133–141. [Google Scholar] [CrossRef]
- Macari, E.R.; Schaeffer, E.K.; West, R.J.; Lowrey, C.H. Simvastatin and t-butylhydroquinone suppress KLF1 and BCL11A gene expression and additively increase fetal hemoglobin in primary human erythroid cells. Blood 2013, 121, 830–839. [Google Scholar] [CrossRef]
- Zhu, X.; Oseghale, A.R.; Nicole, L.H.; Li, B.; Pace, B.S. Mechanisms of NRF2 activation to mediate fetal hemoglobin induction and protection against oxidative stress in sickle cell disease. Exp. Biol. Med. 2019, 244, 171–182. [Google Scholar] [CrossRef]
- Zhu, X.; Li, B.; Pace, B.S. NRF2 mediates gamma-globin gene regulation and fetal hemoglobin induction in human erythroid progenitors. Haematologica 2017, 102, e285–e288. [Google Scholar] [CrossRef]
- Zhu, X.; Xi, C.; Ward, A.; Takezaki, M.; Shi, H.; Peterson, K.R.; Pace, B.S. NRF2 mediates gamma-globin gene regulation through epigenetic modifications in a beta-YAC transgenic mouse model. Exp. Biol. Med. 2020, 245, 1308–1318. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.; Pace, B.; Gupta, D.; Sturtevant, S.; Li, B.; Makala, L.; Brittain, J.; Moore, N.; Vieira, B.F.; Thullen, T.; et al. Dimethyl fumarate increases fetal hemoglobin, provides heme detoxification, and corrects anemia in sickle cell disease. JCI Insight 2017, 2, e96409. [Google Scholar] [CrossRef]
- Hoppe, C.; Jacob, E.; Styles, L.; Kuypers, F.; Larkin, S.; Vichinsky, E. Simvastatin reduces vaso-occlusive pain in sickle cell anaemia: A pilot efficacy trial. Br. J. Haematol. 2017, 177, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Xi, C.; Palani, C.; Takezaki, M.; Shi, H.; Horuzsko, A.; Pace, B.S.; Zhu, X. Simvastatin-Mediated Nrf2 Activation Induces Fetal Hemoglobin and Antioxidant Enzyme Expression to Ameliorate the Phenotype of Sickle Cell Disease. Antioxidants 2024, 13, 337. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.M.; Kasztan, M.; Sedaka, R.; Molina, P.A.; Dunaway, L.S.; Pollock, J.S.; Pollock, D.M. Hydroxyurea improves nitric oxide bioavailability in humanized sickle cell mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2021, 320, R630–R640. [Google Scholar] [CrossRef] [PubMed]
- King, S.B. Nitric oxide production from hydroxyurea. Free Radic. Biol. Med. 2004, 37, 737–744. [Google Scholar] [CrossRef]
- Tshilolo, L.; Tomlinson, G.; Williams, T.N.; Santos, B.; Olupot-Olupot, P.; Lane, A.; Aygun, B.; Stuber, S.E.; Latham, T.S.; McGann, P.T.; et al. Hydroxyurea for Children with Sickle Cell Anemia in Sub-Saharan Africa. N. Engl. J. Med. 2019, 380, 121–131. [Google Scholar] [CrossRef]
- John, C.C.; Opoka, R.O.; Latham, T.S.; Hume, H.A.; Nabaggala, C.; Kasirye, P.; Ndugwa, C.M.; Lane, A.; Ware, R.E. Hydroxyurea Dose Escalation for Sickle Cell Anemia in Sub-Saharan Africa. N. Engl. J. Med. 2020, 382, 2524–2533. [Google Scholar] [CrossRef]
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N. Engl. J. Med. 2018, 379, 226–235. [Google Scholar] [CrossRef]
- Ataga, K.I.; Kutlar, A.; Kanter, J.; Liles, D.; Cancado, R.; Friedrisch, J.; Guthrie, T.H.; Knight-Madden, J.; Alvarez, O.A.; Gordeuk, V.R.; et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 429–439. [Google Scholar] [CrossRef]
- Migotsky, M.; Beestrum, M.; Badawy, S.M. Recent Advances in Sickle-Cell Disease Therapies: A Review of Voxelotor, Crizanlizumab, and L-glutamine. Pharmacy 2022, 10, 123. [Google Scholar] [CrossRef]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N. Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef]
- Frangoul, H.; Locatelli, F.; Sharma, A.; Bhatia, M.; Mapara, M.; Molinari, L.; Wall, D.; Liem, R.I.; Telfer, P.; Shah, A.J.; et al. Exagamglogene Autotemcel for Severe Sickle Cell Disease. N. Engl. J. Med. 2024, 390, 1649–1662. [Google Scholar] [CrossRef] [PubMed]
- Philippidis, A. CASGEVY Makes History as FDA Approves First CRISPR/Cas9 Genome Edited Therapy. Hum. Gene Ther. 2024, 35, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kanter, J.; Walters, M.C.; Krishnamurti, L.; Mapara, M.Y.; Kwiatkowski, J.L.; Rifkin-Zenenberg, S.; Aygun, B.; Kasow, K.A.; Pierciey, F.J., Jr.; Bonner, M.; et al. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. N. Engl. J. Med. 2022, 386, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Herring, W.L.; Gallagher, M.E.; Shah, N.; Morse, K.C.; Mladsi, D.; Dong, O.M.; Chawla, A.; Leiding, J.W.; Zhang, L.; Paramore, C.; et al. Cost-Effectiveness of Lovotibeglogene Autotemcel (Lovo-Cel) Gene Therapy for Patients with Sickle Cell Disease and Recurrent Vaso-Occlusive Events in the United States. Pharmacoeconomics 2024, 42, 693–714. [Google Scholar] [CrossRef]
- Hong, Y.; Boiti, A.; Vallone, D.; Foulkes, N.S. Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution. Antioxidants 2024, 13, 312. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, oxidative stress, and antioxidants: Back and forth in the pathophysiology of chronic diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Haghi Aminjan, H.; Abtahi, S.R.; Hazrati, E.; Chamanara, M.; Jalili, M.; Paknejad, B. Targeting of oxidative stress and inflammation through ROS/NF-kappaB pathway in phosphine-induced hepatotoxicity mitigation. Life Sci. 2019, 232, 116607. [Google Scholar] [CrossRef]
- Veith, A.; Moorthy, B. Role of cytochrome P450s in the generation and metabolism of reactive oxygen species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Stading, R.; Chu, C.; Couroucli, X.; Lingappan, K.; Moorthy, B. Molecular role of cytochrome P4501A enzymes inoxidative stress. Curr. Opin. Toxicol. 2020, 20–21, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Vermot, A.; Petit-Hartlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Cantu-Medellin, N.; Kelley, E.E. Xanthine oxidoreductase-catalyzed reactive species generation: A process in critical need of reevaluation. Redox Biol. 2013, 1, 353–358. [Google Scholar] [CrossRef]
- Tran, N.; Garcia, T.; Aniqa, M.; Ali, S.; Ally, A.; Nauli, S.M. Endothelial nitric oxide synthase (eNOS) and the cardiovascularsystem: In physiology and in disease states. Am. J. Biomed. Sci. Res. 2022, 15, 153–177. [Google Scholar]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Schieber, M.; Navdeep, C.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 1973, 134, 707–716. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef]
- Sharma, L.K.; Lu, J.; Bai, Y. Mitochondrial respiratory complex I: Structure, function and implication in human diseases. Curr. Med. Chem. 2009, 16, 1266–1277. [Google Scholar] [CrossRef]
- Cogliati, S.; Lorenzi, I.; Rigoni, G.; Caicci, F.; Soriano, M.E. Regulation of mitochondrial electron transport chain assembly. J. Mol. Biol. 2018, 430, 4849–4873. [Google Scholar] [CrossRef]
- Bindoli, A.; Rigobello, M.P. Principles in redox signaling: From chemistry to functional significance. Antioxid. Redox Signal. 2013, 18, 1557–1593. [Google Scholar] [CrossRef]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox Signaling Mediated by Thioredoxin and Glutathione Systems in the Central Nervous System. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef]
- Collins, A.R.; Lyon, C.J.; Xia, X.; Liu, J.Z.; Tangirala, R.K.; Yin, F.; Boyadjian, R.; Bikineyeva, A.; Pratico, D.; Harrison, D.G.; et al. Age-accelerated atherosclerosis correlates with failure to upregulate antioxidant genes. Circ. Res. 2009, 104, e42–e54. [Google Scholar] [CrossRef]
- Rasheed, Z. Therapeutic potentials of catalase: Mechanisms, applications, and future perspectives. Int. J. Health Sci. 2024, 18, 1–6. [Google Scholar]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [PubMed]
- Antwi-Boasiako, C.; Dankwah, G.B.; Aryee, R.; Hayfron-Benjamin, C.; Donkor, E.S.; Campbell, A.D. Oxidative Profile of Patients with Sickle Cell Disease. Med. Sci. 2019, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.M.; Liu, M.; Sturdy, M.; Gao, G.; Varghese, S.T.; Sovari, A.A.; Dudley, S.C., Jr. Metabolic stress, reactive oxygen species, and arrhythmia. J. Mol. Cell Cardiol. 2012, 52, 454–463. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Sachdev, V.; Jison, M.L.; Shizukuda, Y.; Plehn, J.F.; Minter, K.; Brown, B.; Coles, W.A.; Nichols, J.S.; Ernst, I.; et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N. Engl. J. Med. 2004, 350, 886–895. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Vichinsky, E. Pulmonary complications of sickle cell disease. N. Engl. J. Med. 2008, 359, 2254–2265. [Google Scholar] [CrossRef]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O., 3rd; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef]
- Conran, N.; Belcher, J.D. Inflammation in sickle cell disease. Clin. Hemorheol. Microcirc. 2018, 68, 263–299. [Google Scholar] [CrossRef]
- Judge, S.; Jang, Y.M.; Smith, A.; Hagen, T.; Leeuwenburgh, C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: Implications for the mitochondrial theory of aging. FASEB J. 2005, 19, 419–421. [Google Scholar] [CrossRef]
- Akhter, M.S.; Hamali, H.A.; Rashid, H.; Dobie, G.; Madkhali, A.M.; Mobarki, A.A.; Oldenburg, J.; Biswas, A. Mitochondria: Emerging Consequential in Sickle Cell Disease. J. Clin. Med. 2023, 12, 765. [Google Scholar] [CrossRef]
- Wood, K.C.; Gladwin, M.T.; Straub, A.C. Sickle cell disease: At the crossroads of pulmonary hypertension and diastolic heart failure. Heart 2020, 106, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Camus, S.M.; De Moraes, J.A.; Bonnin, P.; Abbyad, P.; Le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.C.; Charue, D.; et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 2015, 125, 3805–3814. [Google Scholar] [CrossRef] [PubMed]
- Gbotosho, O.T.; Kapetanaki, M.G.; Kato, G.J. The worst things in life are free: The role of free heme in sickle cell disease. Front. Immunol. 2020, 11, 561917. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Zhang, P.; Abdulla, F.; Nguyen, P.; Killeen, T.; Xu, P.; O’Sullivan, G.; Nath, K.A.; et al. Control of oxidative stress and inflammation in sickle cell disease with the Nrf2 activator dimethyl fumarate. Antioxid. Redox Signal. 2017, 26, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Brousse, V.; Buffet, P.; Rees, D. The spleen and sickle cell disease: The sick(led) spleen. Br. J. Haematol. 2014, 166, 165–176. [Google Scholar] [CrossRef]
- Pizzi, M.; Fuligni, F.; Santoro, L.; Sabattini, E.; Ichino, M.; De Vito, R.; Zucchetta, P.; Colombatti, R.; Sainati, L.; Gamba, P.; et al. Spleen histology in children with sickle cell disease and hereditary spherocytosis: Hints on the disease pathophysiology. Hum. Pathol. 2017, 60, 95–103. [Google Scholar] [CrossRef]
- Ben Khaled, M.; Ouederni, M.; Mankai, Y.; Rekaya, S.; Ben Fraj, I.; Dhouib, N.; Kouki, R.; Mellouli, F.; Bejaoui, M. Prevalence and predictive factors of splenic sequestration crisis among 423 pediatric patients with sickle cell disease in Tunisia. Blood Cells Mol. Dis. 2020, 80, 102374. [Google Scholar] [CrossRef]
- Al-Rimawi, H.S.; Abdul-Qader, M.; Jallad, M.F.; Amarin, Z.O. Acute splenic sequestration in female children with sickle cell disease in the North of Jordan. J. Trop. Pediatr. 2006, 52, 416–420. [Google Scholar] [CrossRef]
- Bandyopadhyay, R.; Bandyopadhyay, S.K.; Dutta, A. Sickle cell hepatopathy. Indian. J. Pathol. Microbiol. 2008, 51, 284–285. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Owen, C.; Chopra, S. Sickle cell hepatopathy. Hepatology 2001, 33, 1021–1028. [Google Scholar] [CrossRef]
- Darbari, D.S.; Kple-Faget, P.; Kwagyan, J.; Rana, S.; Gordeuk, V.R.; Castro, O. Circumstances of death in adult sickle cell disease patients. Am. J. Hematol. 2006, 81, 858–863. [Google Scholar] [CrossRef] [PubMed]
- Hamideh, D.; Alvarez, O. Sickle cell disease related mortality in the United States (1999-2009). Pediatr. Blood Cancer 2013, 60, 1482–1486. [Google Scholar] [CrossRef] [PubMed]
- Lacaille, F.; Allali, S.; de Montalembert, M. The liver in sickle cell disease. J. Pediatr. Gastroenterol. Nutr. 2021, 72, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Badria, F.A.; Ibrahim, A.S.; Badria, A.F.; Elmarakby, A.A. Curcumin attenuates iron accumulation and oxidative stress in the liver and spleen of chronic iron-overloaded rats. PLoS ONE 2015, 10, e0134156. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, H.; Weihrauch, D.; Jones, D.W.; Jing, X.; Shi, Y.; Gourlay, D.; Oldham, K.T.; Hillery, C.A.; Pritchard, K.A., Jr. Inhibition of myeloperoxidase decreases vascular oxidative stress and increases vasodilatation in sickle cell disease mice. J. Lipid Res. 2013, 54, 3009–3015. [Google Scholar] [CrossRef]
- Chou, S.T.; Fasano, R.M. Management of patients with sickle cell disease using transfusion therapy: Guidelines and complications. Hematol. Oncol. Clin. N. Am. 2016, 30, 591–608. [Google Scholar] [CrossRef]
- Kato, G.J.; Hebbel, R.P.; Steinberg, M.H.; Gladwin, M.T. Vasculopathy in sickle cell disease: Biology, pathophysiology, genetics, translational medicine, and new research directions. Am. J. Hematol. 2009, 84, 618–625. [Google Scholar] [CrossRef]
- Do, B.K.; Rodger, D.C. Sickle cell disease and the eye. Curr. Opin. Ophthalmol. 2017, 28, 623–628. [Google Scholar] [CrossRef]
- Beetsch, J.W.; Park, T.S.; Dugan, L.L.; Shah, A.R.; Gidday, J.M. Xanthine oxidase-derived superoxide causes reoxygenation injury of ischemic cerebral endothelial cells. Brain Res. 1998, 786, 89–95. [Google Scholar] [CrossRef]
- Hamer, I.; Wattiaux, R.; Wattiaux-De Coninck, S. Deleterious effects of xanthine oxidase on rat liver endothelial cells after ischemia/reperfusion. Biochim. Biophys. Acta 1995, 1269, 145–152. [Google Scholar] [CrossRef]
- Solovey, A.; Gui, L.; Key, N.S.; Hebbel, R.P. Tissue factor expression by endothelial cells in sickle cell anemia. J. Clin. Investig. 1998, 101, 1899–1904. [Google Scholar] [CrossRef] [PubMed]
- Promsote, W.; Powell, F.L.; Veean, S.; Thounaojam, M.; Markand, S.; Saul, A.; Gutsaeva, D.; Bartoli, M.; Smith, S.B.; Ganapathy, V.; et al. Oral Monomethyl Fumarate Therapy Ameliorates Retinopathy in a Humanized Mouse Model of Sickle Cell Disease. Antioxid. Redox Signal 2016, 25, 921–935. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.J.; Jennette, J.C. Sickle cell nephropathy. Adv. Nephrol. Necker Hosp. 1994, 23, 133–147. [Google Scholar] [PubMed]
- Saborio, P.; Scheinman, J.I. Sickle cell nephropathy. J. Am. Soc. Nephrol. 1999, 10, 187–192. [Google Scholar] [CrossRef]
- Pham, P.T.; Pham, P.C.; Wilkinson, A.H.; Lew, S.Q. Renal abnormalities in sickle cell disease. Kidney Int. 2000, 57, 1–8. [Google Scholar] [CrossRef]
- Hebbel, R.P. The sickle erythrocyte in double jeopardy: Autoxidation and iron decompartmentalization. Semin. Hematol. 1990, 27, 51–69. [Google Scholar]
- Nath, K.A.; Grande, J.P.; Haggard, J.J.; Croatt, A.J.; Katusic, Z.S.; Solovey, A.; Hebbel, R.P. Oxidative stress and induction of heme oxygenase-1 in the kidney in sickle cell disease. Am. J. Pathol. 2001, 158, 893–903. [Google Scholar] [CrossRef]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef]
- Juncos, J.P.; Grande, J.P.; Croatt, A.J.; Hebbel, R.P.; Vercellotti, G.M.; Katusic, Z.S.; Nath, K.A. Early and prominent alterations in hemodynamics, signaling, and gene expression following renal ischemia in sickle cell disease. Am. J. Physiol. Renal Physiol. 2010, 298, F892–F899. [Google Scholar] [CrossRef]
- Jin, Q.; Liu, T.; Qiao, Y.; Liu, D.; Yang, L.; Mao, H.; Ma, F.; Wang, Y.; Peng, L.; Zhan, Y. Oxidative stress and inflammation in diabetic nephropathy: Role of polyphenols. Front. Immunol. 2023, 14, 1185317. [Google Scholar] [CrossRef]
- Goedeke, L.; Fernandez-Hernando, C. Regulation of cholesterol homeostasis. Cell Mol. Life Sci. 2012, 69, 915–930. [Google Scholar] [CrossRef] [PubMed]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.J.; Hong, E.M.; Kim, M.; Kim, J.H.; Jang, J.; Park, S.W.; Byun, H.W.; Koh, D.H.; Choi, M.H.; Kae, S.H.; et al. Simvastatin induces heme oxygenase-1 via NF-E2-related factor 2 (Nrf2) activation through ERK and PI3K/Akt pathway in colon cancer. Oncotarget 2016, 7, 46219–46229. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Feng, Y.; Cui, R.; Qiu, M.; Zhang, J.; Liu, C. Simvastatin protects against acetaminophen-induced liver injury in mice. Biomed. Pharmacother. 2018, 98, 916–924. [Google Scholar] [CrossRef]
- Pritchard, K.A., Jr.; Ou, J.; Ou, Z.; Shi, Y.; Franciosi, J.P.; Signorino, P.; Kaul, S.; Ackland-Berglund, C.; Witte, K.; Holzhauer, S.; et al. Hypoxia-induced acute lung injury in murine models of sickle cell disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 286, L705–L714. [Google Scholar] [CrossRef]
- Ihunnah, C.A.; Ghosh, S.; Hahn, S.; Straub, A.C.; Ofori-Acquah, S.F. Nrf2 Activation With CDDO-Methyl Promotes Beneficial and Deleterious Clinical Effects in Transgenic Mice With Sickle Cell Anemia. Front. Pharmacol. 2022, 13, 880834. [Google Scholar] [CrossRef]
- Chaneiam, N.; Changtam, C.; Mungkongdee, T.; Suthatvoravut, U.; Winichagoon, P.; Vadolas, J.; Suksamrarn, A.; Fucharoen, S.; Svasti, S. A reduced curcuminoid analog as a novel inducer of fetal hemoglobin. Ann. Hematol. 2013, 92, 379–386. [Google Scholar] [CrossRef]
- Macari, E.R.; Lowrey, C.H. Induction of human fetal hemoglobin via the NRF2 antioxidant response signaling pathway. Blood 2011, 117, 5987–5997. [Google Scholar] [CrossRef]
- Hahn, C.K.; Lowrey, C.H. Induction of fetal hemoglobin through enhanced translation efficiency of gamma-globin mRNA. Blood 2014, 124, 2730–2734. [Google Scholar] [CrossRef]
- Chan, K.; Lu, R.; Chang, J.C.; Kan, Y.W. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. USA 1996, 93, 13943–13948. [Google Scholar] [CrossRef]
- Keleku-Lukwete, N.; Suzuki, M.; Otsuki, A.; Tsuchida, K.; Katayama, S.; Hayashi, M.; Naganuma, E.; Moriguchi, T.; Tanabe, O.; Engel, J.D.; et al. Amelioration of inflammation and tissue damage in sickle cell model mice by Nrf2 activation. Proc. Natl. Acad. Sci. USA 2015, 112, 12169–12174. [Google Scholar] [CrossRef] [PubMed]
- Nuamsee, K.; Chuprajob, T.; Pabuprapap, W.; Jintaridth, P.; Munkongdee, T.; Phannasil, P.; Vadolas, J.; Chaichompoo, P.; Suksamrarn, A.; Svasti, S. Trienone analogs of curcuminoids induce fetal hemoglobin synthesis via demethylation at (G)gamma-globin gene promoter. Sci. Rep. 2021, 11, 8552. [Google Scholar] [CrossRef] [PubMed]
- Goel, Y.; Arellano, M.A.; Fouda, R.T.; Garcia, N.R.; Lomeli, R.A.; Kerr, D.; Argueta, D.A.; Gupta, M.; Velasco, G.J.; Prince, R.; et al. Targeting sickle cell pathobiology and pain with novel transdermal curcumin. PNAS Nexus 2025, 4, pgaf053. [Google Scholar] [CrossRef] [PubMed]
- Lopez, N.H.; Li, B.; Palani, C.; Siddaramappa, U.; Takezaki, M.; Xu, H.; Zhi, W.; Pace, B.S. Salubrinal induces fetal hemoglobin expression via the stress-signaling pathway in human sickle erythroid progenitors and sickle cell disease mice. PLoS ONE 2022, 17, e0261799. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, H.; Zhao, M.; Chang, C.; Lu, Q. The Bach Family of Transcription Factors: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2016, 50, 345–356. [Google Scholar] [CrossRef]
- Belcher, J.D.; Nataraja, S.; Abdulla, F.; Zhang, P.; Chen, C.; Nguyen, J.; Ruan, C.; Singh, M.; Demes, S.; Olson, L.; et al. The BACH1 inhibitor ASP8731 inhibits inflammation and vaso-occlusion and induces fetal hemoglobin in sickle cell disease. Front. Med. 2023, 10, 1101501. [Google Scholar] [CrossRef]
- Attucks, O.C.; Jasmer, K.J.; Hannink, M.; Kassis, J.; Zhong, Z.; Gupta, S.; Victory, S.F.; Guzel, M.; Polisetti, D.R.; Andrews, R.; et al. Induction of heme oxygenase I (HMOX1) by HPP-4382: A novel modulator of Bach1 activity. PLoS ONE 2014, 9, e101044. [Google Scholar] [CrossRef]
- Xu, L.; Wu, F.; Yang, L.; Wang, F.; Zhang, T.; Deng, X.; Zhang, X.; Yuan, X.; Yan, Y.; Li, Y.; et al. miR-144/451 inhibits c-Myc to promote erythroid differentiation. FASEB J. 2020, 34, 13194–13210. [Google Scholar] [CrossRef]
- Huang, X.; Chao, R.; Zhang, Y.; Wang, P.; Gong, X.; Liang, D.; Wang, Y. CAP1, a target of miR-144/451, negatively regulates erythroid differentiation and enucleation. J. Cell Mol. Med. 2021, 25, 2377–2389. [Google Scholar] [CrossRef]
- Li, B.; Zhu, X.; Ward, C.M.; Starlard-Davenport, A.; Takezaki, M.; Berry, A.; Ward, A.; Wilder, C.; Neunert, C.; Kutlar, A.; et al. MIR-144-mediated NRF2 gene silencing inhibits fetal hemoglobin expression in sickle cell disease. Exp. Hematol. 2019, 70, 85–96.e5. [Google Scholar] [CrossRef]
- Zhu, X.; Xi, C.; Thomas, B.; Pace, B.S. Loss of NRF2 function exacerbates the pathophysiology of sickle cell disease in a transgenic mouse model. Blood 2018, 131, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Hazra, R.; Ihunnah, C.A.; Weidert, F.; Flage, B.; Ofori-Acquah, S.F. Augmented NRF2 activation protects adult sickle mice from lethal acute chest syndrome. Br. J. Haematol. 2018, 182, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Musicki, B.; Liu, T.; Sezen, S.F.; Burnett, A.L. Targeting NADPH oxidase decreases oxidative stress in the transgenic sickle cell mouse penis. J. Sex. Med. 2012, 9, 1980–1987. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Paul, J.; Wang, Y.; Gupta, M.; Vang, D.; Thompson, S.; Jha, R.; Nguyen, J.; Valverde, Y.; Lamarre, Y.; et al. Heme causes pain in sickle mice via toll-like receptor 4-mediated reactive oxygen species- and endoplasmic reticulum stress-induced glial activation. Antioxid. Redox Signal. 2021, 34, 279–293. [Google Scholar] [CrossRef]
- Gupta, K.B.; Dhiman, M.; Mantha, A.K. Gliadin induced oxidative stress and altered cellular responses in human intestinal cells: An in-vitro study to understand the cross-talk between the transcription factor Nrf-2 and multifunctional APE1 enzyme. J. Biochem. Mol. Toxicol. 2022, 36, e23096. [Google Scholar] [CrossRef]
- Pace, B.S.; Shartava, A.; Pack-Mabien, A.; Mulekar, M.; Ardia, A.; Goodman, S.R. Effects of N-acetylcysteine on dense cell formation in sickle cell disease. Am. J. Hematol. 2003, 73, 26–32. [Google Scholar] [CrossRef]
- Nur, E.; Brandjes, D.P.; Teerlink, T.; Otten, H.M.; Oude Elferink, R.P.; Muskiet, F.; Evers, L.M.; ten Cate, H.; Biemond, B.J.; Duits, A.J.; et al. N-acetylcysteine reduces oxidative stress in sickle cell patients. Ann. Hematol. 2012, 91, 1097–1105. [Google Scholar] [CrossRef]
- Fu, X.; Cate, S.A.; Dominguez, M.; Osborn, W.; Ozpolat, T.; Konkel, B.A.; Chen, J.; Lopez, J.A. Cysteine Disulfides (Cys-ss-X) as Sensitive Plasma Biomarkers of Oxidative Stress. Sci. Rep. 2019, 9, 115–123. [Google Scholar] [CrossRef]
- Biemond, B.J.; Shore, B.J.; Wilson, F.; Jochems, J.; Lindqvist, L.M.; Jung, K.; Gentinetta, T.; Costin, S.; Kato, G.J.; Eleftheriou, P.; et al. A phase 1 study of CSL888 (hemopexin) in adult patients with sickle cell disease. Hemasphere 2023, 7, 15. [Google Scholar] [CrossRef]
- Martins, V.D.; Manfredini, V.; Peralba, M.C.; Benfato, M.S. Alpha-lipoic acid modifies oxidative stress parameters in sickle cell trait subjects and sickle cell patients. Clin. Nutr. 2009, 28, 192–197. [Google Scholar] [CrossRef]
- Liu, L.; Zhu, X.; Yu, A.; Ward, C.M.; Pace, B.S. delta-Aminolevulinate induces fetal hemoglobin expression by enhancing cellular heme biosynthesis. Exp. Biol. Med. 2019, 244, 1220–1232. [Google Scholar] [CrossRef] [PubMed]
- Doss, J.F.; Jonassaint, J.C.; Garrett, M.E.; Ashley-Koch, A.E.; Telen, M.J.; Chi, J.T. Phase 1 Study of a Sulforaphane-Containing Broccoli Sprout Homogenate for Sickle Cell Disease. PLoS ONE 2016, 11, e0152895. [Google Scholar] [CrossRef]
- Vinchi, F.; De Franceschi, L.; Ghigo, A.; Townes, T.; Cimino, J.; Silengo, L.; Hirsch, E.; Altruda, F.; Tolosano, E. Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation 2013, 127, 1317–1329. [Google Scholar] [CrossRef]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Abdulla, F.; Zhang, P.; Nguyen, H.; Nguyen, P.; Killeen, T.; Miescher, S.M.; Brinkman, N.; et al. Haptoglobin and hemopexin inhibit vaso-occlusion and inflammation in murine sickle cell disease: Role of heme oxygenase-1 induction. PLoS ONE 2018, 13, e0196455. [Google Scholar] [CrossRef]
- Buehler, P.W.; Swindle, D.; Pak, D.I.; Ferguson, S.K.; Majka, S.M.; Karoor, V.; Moldovan, R.; Sintas, C.; Black, J.; Gentinetta, T.; et al. Hemopexin dosing improves cardiopulmonary dysfunction in murine sickle cell disease. Free Radic. Biol. Med. 2021, 175, 95–107. [Google Scholar] [CrossRef]
- Gentinetta, T.; Belcher, J.D.; Brugger-Verdon, V.; Adam, J.; Ruthsatz, T.; Bain, J.; Schu, D.; Ventrici, L.; Edler, M.; Lioe, H.; et al. Plasma-derived hemopexin as a candidate therapeutic agent for acute vaso-occlusion in sickle cell disease: Preclinical Evidence. J. Clin. Med. 2022, 11, 630. [Google Scholar] [CrossRef]
- Shore, P.M.; Biemond, B.J.; Kesse-Adu, R.; Wahab, E.; Desai, P.C.; Boucher, A.; Eleftheriou, P.; Rijneveld, A.; De Castro, L.M.; Bergmann, S.; et al. Phase 1 study of the safety and pharmacokinetics of CSL889 (Hemopexin) in adults with SCD. J. Sick. Cell Dis. 2024, 1, yoae002-003. [Google Scholar] [CrossRef]
- Stivala, S.; Gobbato, S.; Bonetti, N.; Camici, G.G.; Luscher, T.F.; Beer, J.H. Dietary alpha-linolenic acid reduces platelet activation and collagen-mediated cell adhesion in sickle cell disease mice. J. Thromb. Haemost. 2022, 20, 375–386. [Google Scholar] [CrossRef]
- Morris, C.R.; Brown, L.A.S.; Reynolds, M.; Dampier, C.D.; Lane, P.A.; Watt, A.; Kumari, P.; Harris, F.; Manoranjithan, S.; Mendis, R.D.; et al. Impact of arginine therapy on mitochondrial function in children with sickle cell disease during vaso-occlusive pain. Blood 2020, 136, 1402–1406. [Google Scholar] [CrossRef]
- Rees, C.A.; Brousseau, D.C.; Cohen, D.M.; Villella, A.; Dampier, C.; Brown, K.; Campbell, A.; Chumpitazi, C.E.; Airewele, G.; Chang, T.; et al. Sickle Cell Disease Treatment with Arginine Therapy (STArT): Study protocol for a phase 3 randomized controlled trial. Trials 2023, 24, 538. [Google Scholar] [CrossRef]
- Panda, H.; Keleku-Lukwete, N.; Kuga, A.; Fuke, N.; Suganuma, H.; Suzuki, M.; Yamamoto, M. Dietary supplementation with sulforaphane attenuates liver damage and heme overload in a sickle cell disease murine model. Exp. Hematol. 2019, 77, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Perrine, S.P.; Ginder, G.D.; Faller, D.V.; Dover, G.H.; Ikuta, T.; Witkowska, H.E.; Cai, S.P.; Vichinsky, E.P.; Olivieri, N.F. A short-term trial of butyrate to stimulate fetal-globin-gene expression in the beta-globin disorders. N. Engl. J. Med. 1993, 328, 81–86. [Google Scholar] [CrossRef]
- Atweh, G.F.; Sutton, M.; Nassif, I.; Boosalis, V.; Dover, G.J.; Wallenstein, S.; Wright, E.; McMahon, L.; Stamatoyannopoulos, G.; Faller, D.V.; et al. Sustained induction of fetal hemoglobin by pulse butyrate therapy in sickle cell disease. Blood 1999, 93, 1790–1797. [Google Scholar] [PubMed]
- Molokie, R.; Lavelle, D.; Gowhari, M.; Pacini, M.; Krauz, L.; Hassan, J.; Ibanez, V.; Ruiz, M.A.; Ng, K.P.; Woost, P.; et al. Oral tetrahydrouridine and decitabine for non-cytotoxic epigenetic gene regulation in sickle cell disease: A randomized phase 1 study. PLoS Med. 2017, 14, e1002382. [Google Scholar] [CrossRef]
- Saunthararajah, Y.; Molokie, R.; Saraf, S.; Sidhwani, S.; Gowhari, M.; Vara, S.; Lavelle, D.; DeSimone, J. Clinical effectiveness of decitabine in severe sickle cell disease. Br. J. Haematol. 2008, 141, 126–129. [Google Scholar] [CrossRef]
- Zhu, X.; Hu, T.; Ho, M.H.; Wang, Y.; Yu, M.; Patel, N.; Pi, W.; Choi, J.H.; Xu, H.; Ganapathy, V.; et al. Hydroxyurea differentially modulates activator and repressors of gamma-globin gene in erythroblasts of responsive and non-responsive patients with sickle cell disease in correlation with Index of Hydroxyurea Responsiveness. Haematologica 2017, 102, 1995–2004. [Google Scholar] [CrossRef]
- Ye, L.; Wang, J.; Tan, Y.; Beyer, A.I.; Xie, F.; Muench, M.O.; Kan, Y.W. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and beta-thalassemia. Proc. Natl. Acad. Sci. USA 2016, 113, 10661–10665. [Google Scholar] [CrossRef]
- Venkatesan, V.; Christopher, A.C.; Rhiel, M.; Azhagiri, M.K.K.; Babu, P.; Walavalkar, K.; Saravanan, B.; Andrieux, G.; Rangaraj, S.; Srinivasan, S.; et al. Editing the core region in HPFH deletions alters fetal and adult globin expression for treatment of beta-hemoglobinopathies. Mol. Ther. Nucleic Acids 2023, 32, 671–688. [Google Scholar] [CrossRef]
- Antoniani, C.; Meneghini, V.; Lattanzi, A.; Felix, T.; Romano, O.; Magrin, E.; Weber, L.; Pavani, G.; El Hoss, S.; Kurita, R.; et al. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human beta-globin locus. Blood 2018, 131, 1960–1973. [Google Scholar] [CrossRef]
- Martyn, G.E.; Wienert, B.; Yang, L.; Shah, M.; Norton, L.J.; Burdach, J.; Kurita, R.; Nakamura, Y.; Pearson, R.C.M.; Funnell, A.P.W.; et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat. Genet. 2018, 50, 498–503. [Google Scholar] [CrossRef]
- Weber, L.; Frati, G.; Felix, T.; Hardouin, G.; Casini, A.; Wollenschlaeger, C.; Meneghini, V.; Masson, C.; De Cian, A.; Chalumeau, A.; et al. Editing a gamma-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci. Adv. 2020, 6, eaay9392. [Google Scholar] [CrossRef] [PubMed]
- Palani, C.D.; Zhu, X.; Alagar, M.; Attucks, O.C.; Pace, B.S. Bach1 inhibitor HPP-D mediates gamma-globin gene activation in sickle erythroid progenitors. Blood Cells Mol. Dis. 2024, 104, 102792. [Google Scholar] [CrossRef] [PubMed]
- Mann, G.E. Nrf2-mediated redox signalling in vascular health and disease. Free Radic. Biol. Med. 2014, 75 (Suppl 1), S1. [Google Scholar] [CrossRef] [PubMed]
- Milan, K.L.; Gayatri, V.; Kriya, K.; Sanjushree, N.; Vishwanathan Palanivel, S.; Anuradha, M.; Ramkumar, K.M. MiR-142-5p mediated Nrf2 dysregulation in gestational diabetes mellitus and its impact on placental angiogenesis. Placenta 2024, 158, 192–199. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, L.; Lu, Y.; Zhang, M.; Zhang, Z.; Wang, K.; Lv, J. Down-regulation of microRNA-142-5p attenuates oxygen-glucose deprivation and reoxygenation-induced neuron injury through up-regulating Nrf2/ARE signaling pathway. Biomed. Pharmacother. 2017, 89, 1187–1195. [Google Scholar] [CrossRef]
- Teimouri, M.; Hosseini, H.; Shabani, M.; Koushki, M.; Noorbakhsh, F.; Meshkani, R. Inhibiting miR-27a and miR-142-5p attenuate nonalcoholic fatty liver disease by regulating Nrf2 signaling pathway. IUBMB Life 2020, 72, 361–372. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Drug | Chemical Structure/DNA Sequence | Mechanism of Action | In Vitro/Preclinical SCD Mice Studies | References |
|---|---|---|---|---|
| Simvastatin | 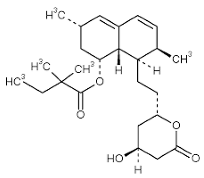 | Competitive 3-hydroxy-3-methyl-glutaryl (HMG)-CoA reductase inhibitor NRF2 agonist, decreases ROS levels. | In vitro and preclinical SCD mice studies NCT01702246. | [107,108] |
| tert-butylhydroquinone | 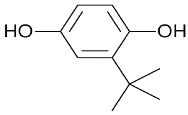 | NRF2 agonist mediating glutathione production. | In vitro. | [109] |
| Dimethyl fumarate | 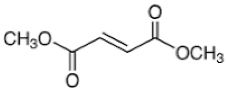 | A small-molecule NRF2 agonist, decreases ROS. | In vitro and preclinical SCD mice studies. | [20,21,110,111] |
| 1-(2-cyano-3,12,28-trioxooleana-1,9(11)-dien-28-yl)-1H-imidazole | 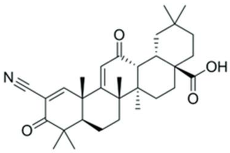 | NRF2 agonist and enhanced ARE binding. | NRF2 agonist, decrease ROS and preclinical SCD mice studies. | [110] [107,112] |
| Curcumin | 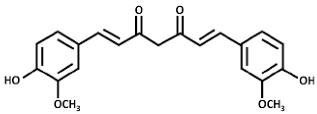 | NRF2 agonist and enhanced ARE binding. | In vitro. | [108,113] [114] |
| Salubrinal | 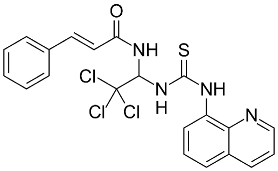 | NRF2 agonist and enhanced ARE binding. | In vitro and preclinical SCD mice. | [115] |
| ASP8731 | 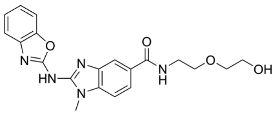 | A selective small molecule inhibitor of BACH1. | In vitro and preclinical SCD mice studies. | [116] |
| HPPD |  | Small molecule inhibitor of BACH1. | In vitro and preclinical SCD mice. | [117,118] |
| miR-144 |  | Inhibits NRF2 through miR-144 target sites in the 3′UTR of NRF2. | In vitro. | [119,120,121] |
| Drug | Chemical Structure | Mechanism of Action | Clinical Trials | References |
|---|---|---|---|---|
| N-acetyl cysteine | 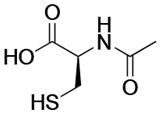 | Precursor for glutathione biosynthesis, decreases ROS. | NCT01800526 NCT01849016 | [127] |
| Hemopexin | 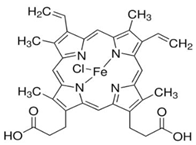 | Decreases ROS by binding to free heme. | NCT06699849 | [130] |
| Alpha-lipoic acid |  | NRF2 agonist that mediates glutathione production. | NCT03161028 | [131] |
| δ-aminolaevulinate | 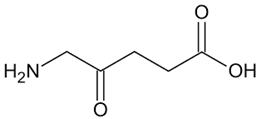 | Metabolic precursor of protoporphyrin IX in heme biosynthesis. | NRF2 agonist and treatment modality for actinic keratosis. | [132] |
| L-Arginine | 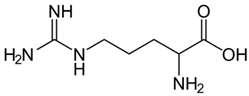 | Precursor for nitic oxide. | Reduce ROS, improved mitochondrial function. NCT04839354 | [107,133] |
| Sulforaphane |  | Increases NRF2 translocation to the nucleus. | NCT01715480 | [133] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Starlard-Davenport, A.; Palani, C.D.; Zhu, X.; Pace, B.S. Innovations in Drug Discovery for Sickle Cell Disease Targeting Oxidative Stress and NRF2 Activation—A Short Review. Int. J. Mol. Sci. 2025, 26, 4192. https://doi.org/10.3390/ijms26094192
Starlard-Davenport A, Palani CD, Zhu X, Pace BS. Innovations in Drug Discovery for Sickle Cell Disease Targeting Oxidative Stress and NRF2 Activation—A Short Review. International Journal of Molecular Sciences. 2025; 26(9):4192. https://doi.org/10.3390/ijms26094192
Chicago/Turabian StyleStarlard-Davenport, Athena, Chithra D. Palani, Xingguo Zhu, and Betty S. Pace. 2025. "Innovations in Drug Discovery for Sickle Cell Disease Targeting Oxidative Stress and NRF2 Activation—A Short Review" International Journal of Molecular Sciences 26, no. 9: 4192. https://doi.org/10.3390/ijms26094192
APA StyleStarlard-Davenport, A., Palani, C. D., Zhu, X., & Pace, B. S. (2025). Innovations in Drug Discovery for Sickle Cell Disease Targeting Oxidative Stress and NRF2 Activation—A Short Review. International Journal of Molecular Sciences, 26(9), 4192. https://doi.org/10.3390/ijms26094192