
Potential Correlation Between Molecular Biomarkers and Oxidative Stress in Traumatic Brain Injury

 ,
,  , , , and
, , , and 
{kind=link}
Abstract
1. Introduction
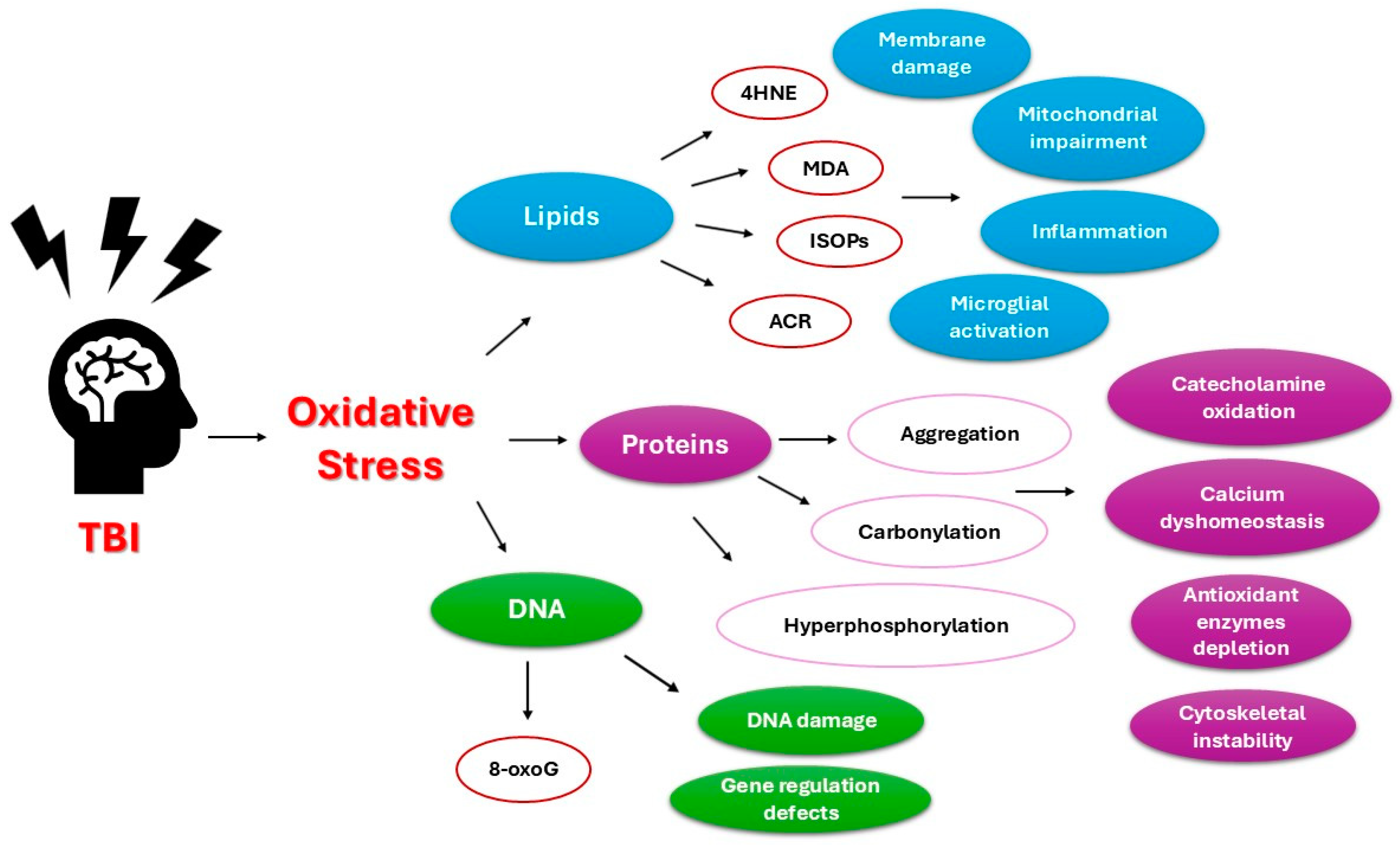
2. Oxidative Stress in Traumatic Brain Injury
3. Antioxidant Defence in Traumatic Brain Injury
4. Neuronal Damage Biomarkers and Oxidative Stress
4.1. Neuron-Specific Enolase
4.2. Ubiquitin C-Terminal Hydrolase L1
4.3. S100 Calcium-Binding Protein B
4.4. Glial Fibrillary Acidic Protein
4.5. Neurofilament Proteins
4.6. Myelin Basic Protein
5. Neurodegeneration Biomarkers and Oxidative Stress
5.1. Tau Protein
5.2. Beta Amyloids
6. Genetic Biomarkers and Oxidative Stress
6.1. Transactive Response DNA-Binding Protein-43
6.2. Micro RNA
6.3. Long Non-Coding RNA
7. Challenges and Future Perspectives
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Galgano, M.; Toshkezi, G.; Qiu, X.; Russell, T.; Chin, L.; Zhao, L.R. Traumatic brain injury: Current treatment strategies and future endeavors. Cell Transplant. 2017, 26, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Lee, A.Y.W. Traumatic brain injuries: Pathophysiology and potential therapeutic targets. Front. Cell. Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef]
- Khellaf, A.; Khan, D.; Helmy, A. Recent advances in traumatic brain injury. J. Neurol. 1019, 266, 2878–2889. [Google Scholar] [CrossRef]
- Silvestro, S.; Raffaele, I.; Quartarone, A.; Mazzon, E. Innovative Insights into Traumatic Brain Injuries: Biomarkers and New Pharmacological Targets. Int. J. Mol. Sci. 2024, 25, 2372. [Google Scholar] [CrossRef] [PubMed]
- Capizzi, A.; Woo, J.; Verduzco-Gutierrez, M. Traumatic Brain Injury: An Overview of Epidemiology, Pathophysiology, and Medical Management. Med. Clin. N. Am. 2020, 104, 213–238. [Google Scholar] [CrossRef]
- Ghaith, H.S.; Nawar, A.A.; Gabra, M.D.; Abdelrahman, M.E.; Nafady, M.H.; Bahbah, E.I.; Ebada, M.A.; Ashraf, G.M.; Negida, A.; Barreto, G.E. A Literature Review of Traumatic Brain Injury Biomarkers. Mol. Neurobiol. 2022, 59, 4141–4158. [Google Scholar] [CrossRef]
- Karimova, D.; Rostami, E.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B.; Rask-Andersen, M. Advances in development of biomarkers for brain damage and ischemia. Mol. Biol. Rep. 2024, 51, 803. [Google Scholar] [CrossRef] [PubMed]
- Prins, M.; Greco, T.; Alexander, D.; Giza, C.C. The pathophysiology of traumatic brain injury at a glance. Dis. Model. Mech. 2013, 6, 1307–1315. [Google Scholar] [CrossRef]
- Cáceres, E.; Olivella, J.C.; Di Napoli, M.; Raihane, A.S.; Divani, A.A. Immune Response in Traumatic Brain Injury. Curr. Neurol. Neurosci. Rep. 2024, 24, 593–609. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, C.Y.; Kim, H.R.; Lee, C.H.; Kim, H.W.; Kim, J.H. A Role of Serum-Based Neuronal and Glial Markers as Potential Predictors for Distinguishing Severity and Related Outcomes in Traumatic Brain Injury. J. Korean Neurosurg. Soc. 2015, 58, 93–100. [Google Scholar] [CrossRef]
- Tong, K.A.; Oyoyo, U.E.; Holshouser, B.A.; Ashwal, S.; Medina, L.S. Traumatic Brain Injury: Evidence-Based Neuroimaging. In Evidence-Based Neuroimaging Diagnosis and Treatment. Evidence-Based Imaging; Medina, L.S., Sanelli, P.C., Jarvik, J.G., Eds.; Springer: New York, NY, USA, 2013. [Google Scholar] [CrossRef]
- Kvietys, P.R.; Granger, N. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef]
- Nguyen, A.; Patel, A.B.; Kioutchoukova, I.P.; Diaz, M.J.; Lucke-Wold, B. Mechanisms of Mitochondrial Oxidative Stress in Brain Injury: From Pathophysiology to Therapeutics. Oxygen 2023, 3, 163–178. [Google Scholar] [CrossRef]
- Mendes Arent, A.; Souza, L.F.D.; Walz, R.; Dafre, A.L. Perspectives on molecular biomarkers of oxidative stress and antioxidant strategies in traumatic brain injury. BioMed Res. Int. 2014, 2014, 723060. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.; Shakkour, Z.; Tabet, M.; Abdelhady, S.; Kobaisi, A.; Abedi, R.; Nasrallah, L.; Pintus, G.; Al-Dhaheri, Y.; Mondello, S.; et al. Traumatic Brain Injury: Oxidative Stress and Novel Anti-Oxidants Such as Mitoquinone and Edaravone. Antioxidants 2020, 9, 943. [Google Scholar] [CrossRef]
- Abdul-Muneer, P.M.; Chandra, N.; Haorah, J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol. Neurobiol. 2015, 51, 966–979. [Google Scholar] [CrossRef] [PubMed]
- Breitzig, M.; Bhimineni, C.; Lockey, R.; Kolliputi, N. 4-Hydroxy-2-nonenal: A critical target in oxidative stress? Am. J. Physiol. Cell Physiol. 2016, 311, C537–C543. [Google Scholar] [CrossRef] [PubMed]
- Milatovic, D.; Aschner, M. Measurement of isoprostanes as markers of oxidative stress in neuronal tissue. Curr. Protoc. Toxicol. 2009, 39, 12–14. [Google Scholar] [CrossRef]
- Lorente, L.; Martín, M.M.; Abreu-González, P.; Ramos, L.; Argueso, M.; Cáceres, J.J.; Sole-Violan, J.; Lorenzo, J.M.; Molina, I.; Jiménez, A. Association between serum malondialdehyde levels and mortality in patients with severe brain trauma injury. J. Neurotrauma 2015, 32, 1–6. [Google Scholar] [CrossRef]
- Thapak, P.; Gomez-Pinilla, F. The bioenergetics of traumatic brain injury and its long-term impact for brain plasticity and function. Pharmacol. Res. 2024, 208, 107389. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Garbarino, V.R.; Orr, M.E.; Rodriguez, K.A.; Buffenstein, R. Mechanisms of oxidative stress resistance in the brain: Lessons learned from hypoxia tolerant extremophilic vertebrates. Arch. Biochem. Biophys. 2015, 576, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef] [PubMed]
- Fehily, B.; Fitzgerald, M. Repeated mild traumatic brain injury: Potential mechanisms of damage. Cell Transplant. 2017, 26, 1131–1155. [Google Scholar] [CrossRef]
- Gruener, N.; Gross, B.; Gozlan, O.; Barak, M. Increase in superoxide dismutase after cerebrovascular accident. Life Sci. 1994, 54, 711–713. [Google Scholar] [CrossRef]
- Loane, D.J.; Kumar, A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 2016, 275 Pt 3, 316–327. [Google Scholar] [CrossRef]
- Cai, Z.; Zhao, B.; Ratka, A. Oxidative Stress and β-Amyloid Protein in Alzheimer’s Disease. Neuromolecular Med. 2011, 13, 223–250. [Google Scholar] [CrossRef]
- Loane, D.J.; Kumar, A.; Stoica, B.A.; Cabatbat, R.; Faden, A.I. Progressive neurodegeneration after experimental brain trauma: Association with chronic microglial activation. J. Neuropathol. Exp. Neurol. 2014, 73, 14–29. [Google Scholar] [CrossRef]
- Gélinas, S.; Chapados, C.; Beauregard, M.; Gosselin, I.; Martinoli, M.G. Effect of oxidative stress on stability and structure of neurofilament proteins. Biochem. Cell Biol. 2000, 78, 667–674. [Google Scholar] [CrossRef]
- Castellani, R.J.; Perry, G. Tau biology, tauopathy, traumatic brain injury, and diagnostic challenges. J. Alzheimer’s Dis. 2019, 67, 447–467. [Google Scholar] [CrossRef]
- Uryu, K.; Laurer, H.; McIntosh, T.; Praticò, D.; Martinez, D.; Leight, S.; Lee, V.M.; Trojanowski, J.Q. Repetitive mild brain trauma accelerates Abeta deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J. Neurosci. 2002, 22, 446–454. [Google Scholar] [CrossRef]
- Abdul-Muneer, P.M. Nrf2 as a Potential Therapeutic Target for Traumatic Brain Injury. J. Integr. Neurosci. 2023, 22, 81. [Google Scholar] [CrossRef]
- Fesharaki-Zadeh, A. Oxidative Stress in Traumatic Brain Injury. Int. J. Mol. Sci. 2022, 23, 13000. [Google Scholar] [CrossRef]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Yunoki, M.; Kawauchi, M.; Ukita, N.; Noguchi, Y.; Nishio, S.; Ono, Y.; Asari, S.; Ohmoto, T.; Asanuma, M.; Ogawa, N. Effects of lecithinized superoxide dismutase on traumatic brain injury in rats. J. Neurotrauma 1997, 14, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Nandi, A.; Yan, L.J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxid Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef] [PubMed]
- Sackheim, A.M.; Villalba, N.; Bonev, A.; Nelson, M.; Freeman, K. Increased Hydrogen Peroxide After Traumatic Brain Injury Disrupts Phosphatidylinositol 4,5-Bisphosphate Metabolism Causing Impaired Inward Rectifier Potassium Channel Function. FASEB J. 2018, 32, 703.4. [Google Scholar] [CrossRef]
- Li, J.; Li, W.; Su, J.; Liu, W.; Altura, B.T.; Altura, B.M. Hydrogen peroxide induces apoptosis in cerebral vascular smooth muscle cells: Possible relation to neurodegenerative diseases and strokes. Brain Res Bull. 2003, 62, 101–106. [Google Scholar] [CrossRef]
- Lutton, E.M.; Farney, S.K.; Andrews, A.M.; Shuvaev, V.V.; Chuang, G.Y.; Muzykantov, V.R.; Ramirez, S.H. Endothelial targeted strategies to combat oxidative stress: Improving outcomes in traumatic brain injury. Front. Neurol. 2019, 10, 582. [Google Scholar] [CrossRef]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxidants Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef]
- Fan, P.; Yamauchi, T.; Noble, L.J.; Ferriero, D.M. Age-dependent differences in glutathione peroxidase activity after traumatic brain injury. J. Neurotrauma 2003, 20, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, V.; Yakoub, K.M.; Caruso, G.; Lazzarino, G.; Signoretti, S.; Barbey, A.K.; Tavazzi, B.; Lazzarino, G.; Belli, A.; Amorini, A.M. Antioxidant therapies in traumatic brain injury. Antioxidants 2020, 9, 260. [Google Scholar] [CrossRef]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Vitamin E protects against oxidative damage and learning disability after mild traumatic brain injury in rats. Neurorehabilit. Neural repair 2010, 24, 290–298. [Google Scholar] [CrossRef]
- Feng, J.; Zheng, Y.; Guo, M.; Ares, I.; Martínez, M.; Lopez-Torres, B.; Martínez-Larrañaga, M.R.; Wang, X.; Anadón, A.; Martínez, M.A. Oxidative stress, the blood-brain barrier and neurodegenerative diseases: The critical beneficial role of dietary antioxidants. Acta Pharm. Sin. B 2023, 13, 3988–4024. [Google Scholar] [CrossRef]
- Glushakova, O.Y.; Glushakov, A.V.; Mannix, R.; Miller, E.R.; Valadka, A.B.; Hayes, R.L. The Use of Blood-Based Biomarkers to Improve the Design of Clinical Trials of Traumatic Brain Injury. In Chapter 8 in Handbook of Neuroemergency Clinical Trials; Academic Press: Cambridge, MA, USA, 2018; pp. 139–166. [Google Scholar] [CrossRef]
- Gordillo-Escobar, E.; Egea-Guerrero, J.J.; Rodríguez-Rodríguez, A.; Murillo-Cabezas, F. Usefulness of biomarkers in the prognosis of severe head injuries. Med. Intensiv. 2016, 40, 105–112. [Google Scholar] [CrossRef]
- Haque, A.; Polcyn, R.; Matzelle, D.; Banik, N.L. New Insights into the Role of Neuron-Specific Enolase in Neuro-Inflammation, Neurodegeneration, and Neuroprotection. Brain Sci. 2018, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.M.; Luo, Y.L.; Li, S.; Li, Z.X.; Jiang, L.; Zhang, G.X.; Owusu, L.; Chen, H.L. Multifunctional neuron-specific enolase: Its role in lung diseases. Biosci. Rep. 2019, 39, BSR20192732. [Google Scholar] [CrossRef]
- Horvat, S.; Kos, J.; Pišlar, A. Multifunctional roles of γ-enolase in the central nervous system: More than a neuronal marker. Cell Biosci. 2024, 14, 61. [Google Scholar] [CrossRef] [PubMed]
- Žurek, J. Biomarkers in Traumatic Brain Injury. In Chapter 33 in Essentials of Neuroanesthesia; Prabhakar, H., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 587–591. [Google Scholar] [CrossRef]
- Haque, A.; Ray, S.K.; Cox, A.; Banik, N.L. Neuron specific enolase: A promising therapeutic target in acute spinal cord injury. Metab. Brain Dis. 2016, 31, 87–95. [Google Scholar] [CrossRef]
- Sogut, O.; Guloglu, C.; Orak, M.; Sayhan, M.B.; Gokdemir, M.T.; Ustundag, M.; Akkus, Z. Trauma scores and neuron-specific enolase, cytokine and C-reactive protein levels as predictors of mortality in patients with blunt head trauma. J. Int. Med. Res. 2010, 38, 1708–1720. [Google Scholar] [CrossRef]
- Meric, E.; Gunduz, A.; Turedi, S.; Cakir, E.; Yandi, M. The prognostic value of neuron-specific enolase in head trauma patients. J. Emerg. Med. 2010, 38, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Lima, J.E.; Takayanagui, O.M.; Garcia, L.V.; Leite, J.P. Use of neuron-specific enolase for assessing the severity and outcome of neurological disorders in patients. Braz. J. Med. Biol. Res. 2004, 37, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Yuan, Q.; Yang, J.; Wang, W.; Liu, H. The prognostic value of serum neuron-specific enolase in traumatic brain injury: Systematic review and meta-analysis. PLoS ONE 2014, 9, e106680. [Google Scholar] [CrossRef]
- Nakhjavan-Shahraki, B.; Yousefifard, M.; Oraii, A.; Sarveazad, A.; Hosseini, M. Meta-analysis of neuron specific enolase in predicting pediatric brain injury outcomes. EXCLI J. 2017, 16, 995. [Google Scholar] [PubMed]
- Isgrò, M.A.; Bottoni, P.; Scatena, R. Neuron-Specific Enolase as a Biomarker: Biochemical and Clinical Aspects. Adv. Exp. Med. Biol. 2015, 867, 125–143. [Google Scholar] [CrossRef]
- Mercier, E.; Tardif, P.A.; Cameron, P.A.; Émond, M.; Moore, L.; Mitra, B.; Ouellet, M.C.; Frenette, J.; de Guise, E.; Le Sage, N. Prognostic value of neuron-specific enolase (NSE) for prediction of post-concussion symptoms following a mild traumatic brain injury: A systematic review. Brain Inj. 2018, 32, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Vasiljevic, B.; Maglajlic-Djukic, S.; Gojnic, M.; Stankovic, S.; Ignjatovic, S.; Lutovac, D. New insights in the pathogenesis of perinatal hypoxic-ischemic brain injury. Pediatr. Int. 2011, 53, 454–462. [Google Scholar] [CrossRef]
- Woertgen, C.; Rothoerl, R.D.; Brawanski, A. Time profile of neuron specific enolase serum levels after experimental brain injury in rat. Acta Neurochir. Suppl. 2000, 76, 371–373. [Google Scholar] [CrossRef]
- Berger, R.P.; Pierce, M.C.; Wisniewski, S.R.; Adelson, P.D.; Clark, R.S.B.; Ruppel, R.A.; Kochanek, P.M. Neuron-Specific Enolase and S100B in Cerebrospinal Fluid After Severe Traumatic Brain Injury in Infants and Children. Pediatrics 2002, 109, e31. [Google Scholar] [CrossRef]
- El-Maraghi, S.; Yehia, H.; Hossam, H.; Yehia, A.; Mowafy, H. The prognostic value of neuron specific enolase in head injury. Egypt. J. Crit. Care Med. 2013, 1, 25–32. [Google Scholar] [CrossRef]
- Wang, K.K.; Munoz-Pareja, J.C.; Lautenslager, L.A.; Tyndall, J.A.; Yang, Z.; Kerrigan, M.R.; Diaz-Arrastia, R.; Korley, F.K.; Okonkwo, D.; Puccio, A.M.; et al. Diagnostic performance of point-of-care ubiquitin carboxy-terminal Hydrolase-L1 assay in distinguishing imaging abnormalities in traumatic brain injury: A TRACK-TBI cohort study. Adv. Biomark. Sci. Technol. 2023, 5, 38–49. [Google Scholar] [CrossRef]
- Yoshida, E.; Mokuno, K.; Aoki, S.; Takahashi, A.; Riku, S.; Murayama, T.; Yanagi, T.; Kato, K. Cerebrospinal fluid levels of superoxide dismutases in neurological diseases detected by sensitive enzyme immunoassays. J. Neurol. Sci. 1994, 124, 25–31. [Google Scholar] [CrossRef]
- Dincel, G.C.; Atmaca, H.T. Role of oxidative stress in the pathophysiology of Toxoplasma gondii infection. Int. J. Immunopathol. Pharmacol. 2016, 29, 226–240. [Google Scholar] [CrossRef]
- Bishop, P.; Rocca, D.; Henley, J.M. Ubiquitin C-terminal hydrolase L1 (UCH-L1): Structure, distribution and roles in brain function and dysfunction. Biochem. J. 2016, 473, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Graham, S. Modification of ubiquitin C-terminal hydrolase L1 by reactive lipid species: Role in neural regeneration and diseases of aging. Neural Regen. Res. 2016, 11, 908–909. [Google Scholar] [CrossRef] [PubMed]
- Brophy, G.M.; Mondello, S.; Papa, L.; Robicsek, S.A.; Gabrielli, A.; Tepas III, J.; Buki, A.; Robertson, C.; Tortella, F.C.; Hayes, R.L.; et al. Biokinetic analysis of ubiquitin C-terminal hydrolase-L1 (UCH-L1) in severe traumatic brain injury patient biofluids. J. Neurotrauma 2011, 28, 861–870. [Google Scholar] [CrossRef]
- Diaz-Arrastia, R.; Wang, K.K.; Papa, L.; Sorani, M.D.; Yue, J.K.; Puccio, A.M.; McMahon, P.J.; Inoue, T.; Yuh, E.L.; Lingsma, H.F.; et al. Acute biomarkers of traumatic brain injury: Relationship between plasma levels of ubiquitin C-terminal hydrolase-L1 and glial fibrillary acidic protein. J. Neurotrauma 2014, 31, 19–25. [Google Scholar] [CrossRef]
- Papa, L.; McKinley, W.I.; Valadka, A.B.; Newman, Z.C.; Nordgren, R.K.; Pramuka, P.E.; Barbosa, C.E.; Brito, A.M.P.; Loss, L.J.; Tinoco-Garcia, L.; et al. Diagnostic Performance of GFAP, UCH-L1, and MAP-2 Within 30 and 60 Minutes of Traumatic Brain Injury. JAMA Netw. Open 2024, 7, e2431115. [Google Scholar] [CrossRef]
- Morris, M.C.; Bercz, A.; Niziolek, G.M.; Kassam, F.; Veile, R.; Friend, L.A.; Pritts, T.A.; Makley, A.T.; Goodman, M.D. UCH-L1 is a Poor Serum Biomarker of Murine Traumatic Brain Injury After Polytrauma. J. Surg. Res. 2019, 244, 63–68. [Google Scholar] [CrossRef]
- Deng, J.; Sun, X.; Liu, K. Relationship between GFAP, UCH-L1 and CT findings and outcome in patients with severe traumatic brain injury. Chongqing Med. 2013, 36, 4117–4119. [Google Scholar]
- Matuszczak, E.; Tylicka, M.; Komarowska, M.D.; Debek, W.; Hermanowicz, A. Ubiquitin carboxy-terminal hydrolase L1—Physiology and pathology. Cell Biochem. Funct. 2020, 38, 533–540. [Google Scholar] [CrossRef]
- Mehta, T.; Fayyaz, M.; Giler, G.E.; Kaur, H.; Raikwar, S.P.; Kempuraj, D.; Selvakumar, G.P.; Ahmed, M.E.; Thangavel, R.; Zaheer, S.; et al. Current Trends in Biomarkers for Traumatic Brain Injury. Open Access J. Neurol. Neurosurg. 2020, 12, 86–94. [Google Scholar]
- Wang, K.K.W.; Kobeissy, F.H.; Shakkour, Z.; Tyndall, J.A. Thorough overview of ubiquitin C-terminal hydrolase-L1 and glial fibrillary acidic protein as tandem biomarkers recently cleared by US Food and Drug Administration for the evaluation of intracranial injuries among patients with traumatic brain injury. Acute Med. Surg. 2021, 8, e622. [Google Scholar] [CrossRef]
- Memari, B.; Bouttier, M.; Dimitrov, V.; Ouellette, M.; Behr, M.A.; Fritz, J.H.; White, J.H. Engagement of the Aryl Hydrocarbon Receptor in Mycobacterium tuberculosis-Infected Macrophages Has Pleiotropic Effects on Innate Immune Signaling. J. Immunol. 2015, 195, 4479–4491. [Google Scholar] [CrossRef] [PubMed]
- Sorci, G.; Riuzzi, F.; Arcuri, C.; Tubaro, C.; Bianchi, R.; Giambanco, I.; Donato, R. S100B protein in tissue development, repair and regeneration. World J. Biol. Chem. 2013, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Failla, M.D.; Niyonkuru, C.; Amin, K.; Fabio, A.; Berger, R.P.; Wagner, A.K. S100b as a prognostic biomarker in outcome prediction for patients with severe traumatic brain injury. J. Neurotrauma 2013, 30, 946–957. [Google Scholar] [CrossRef]
- Thelin, E.P.; Nelson, D.W.; Bellander, B.M. A review of the clinical utility of serum S100B protein levels in the assessment of traumatic brain injury. Acta Neurochir. 2017, 159, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Oris, C.; Pereira, B.; Durif, J.; Simon-Pimmel, J.; Castellani, C.; Manzano, S.; Sapin, V.; Bouvier, D. The Biomarker S100B and Mild Traumatic Brain Injury: A Meta-analysis. Pediatrics 2018, 141, e20180037. [Google Scholar] [CrossRef]
- Filippidis, A.S.; Papadopoulos, D.C.; Kapsalaki, E.Z.; Fountas, K.N. Role of the S100B serum biomarker in the treatment of children suffering from mild traumatic brain injury. Neurosurg. Focus 2010, 29, E2. [Google Scholar] [CrossRef]
- Janigro, D.; Mondello, S.; Posti, J.P.; Unden, J. GFAP and S100B: What You Always Wanted to Know and Never Dared to Ask. Front. Neurol. 2022, 13, 835597. [Google Scholar] [CrossRef]
- Gayger-Dias, V.; Vizuete, A.F.; Rodrigues, L.; Wartchow, K.M.; Bobermin, L.; Leite, M.C.; Quincozes-Santos, A.; Kleindienst, A.; Gonçalves, C.A. How S100B crosses brain barriers and why it is considered a peripheral marker of brain injury. Exp. Biol. Med. 2023, 248, 2109–2119. [Google Scholar] [CrossRef]
- Hermann, A.; Donato, R.; Weiger, T.M.; Chazin, W.J. S100 calcium binding proteins and ion channels. Front. Pharmacol. 2012, 3, 67. [Google Scholar] [CrossRef]
- Unden, J.; Bellner, J.; Eneroth, M.; Alling, C.; Ingebrigtsen, T.; Romner, B. Raised serum S100B levels after acute bone fractures without cerebral injury. J. Trauma Acute Care Surg. 2005, 58, 59–61. [Google Scholar] [CrossRef]
- Oris, C.; Kahouadji, S.; Durif, J.; Bouvier, D.; Sapin, V. S100B, Actor and Biomarker of Mild Traumatic Brain Injury. Int. J. Mol. Sci. 2023, 24, 6602. [Google Scholar] [CrossRef]
- Verduzco-Mendoza, A.; Carrillo-Mora, P.; Avila-Luna, A.; Gálvez-Rosas, A.; Olmos-Hernández, A.; Mota-Rojas, D.; Bueno-Nava, A. Role of the Dopaminergic System in the Striatum and its Association with Functional Recovery or Rehabilitation After Brain Injury. Front. Neurosci. 2021, 15, 693404. [Google Scholar] [CrossRef] [PubMed]
- Ercole, A.; Thelin, E.P.; Holst, A.; Bellander, B.M.; Nelson, D.W. Kinetic modelling of serum S100b after traumatic brain injury. BMC Neurol. 2016, 16, 93. [Google Scholar] [CrossRef] [PubMed]
- Rainey, T.; Lesko, M.; Sacho, R.; Lecky, F.; Childs, C. Predicting outcome after severe traumatic brain injury using the serum S100B biomarker: Results using a single (24h) time-point. Resuscitation 2009, 80, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Seidenfaden, S.C.; Kjerulff, J.L.; Juul, N.; Kirkegaard, H.; Fogh Møller, M.; Bloch Münster, A.M.; Thingemann Bøtker, M. Temporal Changes in Serum S100B Levels From Prehospital to Early In-Hospital Sampling in Patients Suffering Traumatic Brain Injury. Front. Neurol. 2022, 13, 800015. [Google Scholar] [CrossRef]
- Haselmann, V.; Schamberger, C.; Trifonova, F.; Ast, V.; Froelich, M.F.; Strauß, M.; Kittel, M.; Jaruschewski, S.; Eschmann, D.; Neumaier, M.; et al. Plasma-based S100B testing for management of traumatic brain injury in emergency setting. Pract. Lab. Med. 2021, 26, e00236. [Google Scholar] [CrossRef]
- Jolly, A.E.; Raymont, V.; Cole, J.H.; Whittington, A.; Scott, G.; De Simoni, S.; Searle, G.; Gunn, R.N.; Sharp, D.J. Dopamine D2/D3 receptor abnormalities after traumatic brain injury and their relationship to post-traumatic depression. NeuroImage Clin. 2019, 24, 101950. [Google Scholar] [CrossRef]
- Jenkins, P.O.; De Simoni, S.; Bourke, N.J.; Fleminger, J.; Scott, G.; Towey, D.J.; Svensson, W.; Khan, S.; Patel, M.; Greenwood, R.; et al. Dopaminergic abnormalities following traumatic brain injury. Brain 2018, 141, 797–810. [Google Scholar] [CrossRef]
- Mata-Bermudez, A.; Trejo-Chávez, R.; Martínez-Vargas, M.; Pérez-Arredondo, A.; Martínez-Cardenas, M.L.Á.; Diaz-Ruiz, A.; Rios, C.; Navarro, L. Dysregulation of the dopaminergic system secondary to traumatic brain injury: Implications for mood and anxiety disorders. Front. Neurosci. 2024, 18, 1447688. [Google Scholar] [CrossRef] [PubMed]
- Michetti, F.; Clementi, M.E.; Di Liddo, R.; Valeriani, F.; Ria, F.; Rende, M.; Di Sante, G.; Romano Spica, V. The S100B Protein: A Multifaceted Pathogenic Factor More Than a Biomarker. Int. J. Mol. Sci. 2023, 24, 9605. [Google Scholar] [CrossRef] [PubMed]
- Gomes, F.C.A.; Paulin, D.; Moura Neto, V. Glial fibrillary acidic protein (GFAP): Modulation by growth factors and its implication in astrocyte differentiation. Braz. J. Med. Biol. Res. 1999, 32, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Huebschmann, N.A.; Luoto, T.M.; Karr, J.E.; Berghem, K.; Blennow, K.; Zetterberg, H.; Ashton, N.J.; Simren, J.; Posto, J.P.; Gill, J.M.; et al. Comparing glial fibrillary acidic protein (GFAP) in serum and plasma following mild traumatic brain injury in older adults. Front. Neurol. 2020, 11, 1054. [Google Scholar] [CrossRef]
- Lei, J.; Gao, G.; Feng, J.; Jin, Y.; Wang, C.; Mao, Q.; Jiang, J. Glial fibrillary acidic protein as a biomarker in severe traumatic brain injury patients: A prospective cohort study. Crit. Care 2015, 19, 362. [Google Scholar] [CrossRef]
- Papa, L.; Lewis, L.M.; Falk, J.L.; Zhang, Z.; Silvestri, S.; Giordano, P.; Brophy, G.M.; Demery, J.A.; Dixit, N.K.; Ferguson, I.; et al. Elevated levels of serum glial fibrillary acidic protein breakdown products in mild and moderate traumatic brain injury are associated with intracranial lesions and neurosurgical intervention. Ann. Emerg. Med. 2012, 59, 471–483. [Google Scholar] [CrossRef]
- Luoto, T.M.; Raj, R.; Posti, J.P.; Gardner, A.J.; Panenka, W.J.; Iverson, G.L. A systematic review of the usefulness of glial fibrillary acidic protein for predicting acute intracranial lesions following head trauma. Front. Neurol. 2017, 8, 652. [Google Scholar] [CrossRef]
- Papa, L.; Brophy, G.M.; Welch, R.D.; Lewis, L.M.; Braga, C.F.; Tan, C.N.; Ameli, N.J.; Lopez, M.A.; Haeussler, C.A.; Giordano, D.I.M.; et al. Time Course and Diagnostic Accuracy of Glial and Neuronal Blood Biomarkers GFAP and UCH-L1 in a Large Cohort of Trauma Patients With and Without Mild Traumatic Brain Injury. JAMA Neurol. 2016, 73, 551–560. [Google Scholar] [CrossRef]
- Korley, F.K.; Jain, S.; Sun, X.; Puccio, A.M.; Yue, J.K.; Gardner, R.C.; Wang, K.K.W.; O Okonkwo, D.; Yuh, E.L.; Mukherjee, P.; et al. Prognostic value of day-of-injury plasma GFAP and UCH-L1 concentrations for predicting functional recovery after traumatic brain injury in patients from the US TRACK-TBI cohort: An observational cohort study. Lancet Neurol. 2022, 21, 803–813. [Google Scholar] [CrossRef]
- Zwirner, J.; Lier, J.; Franke, H.; Hammer, N.; Matschke, J.; Trautz, F.; Tse, R.; Ondruschka, B. GFAP positivity in neurons following traumatic brain injuries. Int. J. Leg. Med. 2021, 135, 2323–2333. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.E.; Rozovsky, I.; Goldsmith, S.K.; Stone, D.J.; Yoshida, T.; Finch, C.E. Increased transcription of the astrocyte gene GFAP during middle-age is attenuated by food restriction: Implications for the role of oxidative stress. Free Radic. Biol. Med. 1997, 23, 524–528. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, X.; Chen, L.; Lenahan, C.; Fu, Z.; Fang, Y.; Yu, W. Crosstalk Between the Oxidative Stress and Glia Cells After Stroke: From Mechanism to Therapies. Front. Immunol. 2022, 13, 852416. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.; Rao, M.V.; Veeranna, E.; Nixon, R.A. Neurofilaments and Neurofilament Proteins in Health and Disease. Cold Spring Harb. Perspect. Biol. 2017, 9, a018309. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wu, M.; Zhan, C.; Ma, E.; Yang, M.; Yang, X.; Li, Y. Neurofilament proteins in axonal regeneration and neurodegenerative diseases. Neural Regen. Res. 2012, 7, 620–626. [Google Scholar] [CrossRef]
- Yuan, A.; Nixon, R.A. Neurofilament proteins as biomarkers to monitor neurological diseases and the efficacy of therapies. Front. Neurosci. 2021, 15, 689938. [Google Scholar] [CrossRef]
- Wong, K.R.; O’brien, W.T.; Sun, M.; Yamakawa, G.; O’brien, T.J.; Mychasiuk, R.; Shultz, S.R.; McDonald, S.J.; Brady, R.D. Serum Neurofilament Light as a Biomarker of Traumatic Brain Injury in the Presence of Concomitant Peripheral Injury. Biomark. Insights 2021, 16, 11772719211053449. [Google Scholar] [CrossRef]
- Gao, W.; Zhang, Z.; Lv, X.; Wu, Q.; Yan, J.; Mao, G.; Xing, W. Neurofilament light chain level in traumatic brain injury: A system review and meta-analysis. Medicine 2020, 99, e22363. [Google Scholar] [CrossRef]
- Shahim, P.; Gren, M.; Liman, V.; Andreasson, U.; Norgren, N.; Tegner, Y.; Mattsson, N.; Andreasen, N.; Ost, M.; Zetterberg, H.; et al. Serum neurofilament light protein predicts clinical outcome in traumatic brain injury. Sci. Rep. 2016, 6, 36791. [Google Scholar] [CrossRef]
- Graham, N.S.N.; Zimmerman, K.A.; Moro, F.; Heslegrave, A.; Maillard, S.A.; Bernini, A.; Miroz, J.-P.; Donat, C.K.; Lopez, M.Y.; Bourke, N.; et al. Axonal marker neurofilament light predicts long-term outcomes and progressive neurodegeneration after traumatic brain injury. Sci. Transl. Med. 2021, 13, eabg9922. [Google Scholar] [CrossRef]
- Andersson, E.; Öst, M.; Dalla, K.; Zetterberg, H.; Blennow, K.; Nellgård, B. Acute-Phase Neurofilament Light and Glial Fibrillary Acidic Proteins in Cerebrospinal Fluid Predict Long-Term Outcome After Severe Traumatic Brain Injury. Neurocrit. Care 2024, 41, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Newcombe, V.F.J.; Ashton, N.J.; Posti, J.P.; Glocker, B.; Manktelow, A.; A Chatfield, D.; Winzeck, S.; Needham, E.; Correia, M.M.; Williams, G.B.; et al. Post-acute blood biomarkers and disease progression in traumatic brain injury. Brain 2022, 145, 2064–2076. [Google Scholar] [CrossRef]
- Shahim, P.; Politis, A.; van der Merwe, A.; Moore, B.; Chou, Y.-Y.; Pham, D.L.; Butman, J.A.; Diaz-Arrastia, R.; Gill, J.M.; Brody, D.L.; et al. Neurofilament light as a biomarker in traumatic brain injury. Neurology 2020, 95, e610–e622. [Google Scholar] [CrossRef] [PubMed]
- Leski, M.L.; Bao, F.; Wu, L.; Qian, H.; Sun, D.; Liu, D. Protein and DNA oxidation in spinal injury: Neurofilaments--an oxidation target. Free Radic. Biol. Med. 2001, 30, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.G.; Yong, Y.Y.; Pan, Y.R.; Zhang, L.; Wu, J.M.; Zhang, Y.; Tang, Y.; Wei, J.; Yu, L.; Law, B.Y.; et al. Targeting Nrf2-Mediated Oxidative Stress Response in Traumatic Brain Injury: Therapeutic Perspectives of Phytochemicals. Oxidative Med. Cell. Longev. 2022, 2022, 1015791. [Google Scholar] [CrossRef]
- Endale, H.T.; Tesfaye, W.; Mengstie, T.A. ROS induced lipid peroxidation and their role in ferroptosis. Front. Cell Dev. Biol. 2023, 11, 1226044. [Google Scholar] [CrossRef]
- Gowthami, N.; Pursotham, N.; Dey, G.; Ghose, V.; Sathe, G.; Pruthi, N.; Shukla, D.; Gayathri, N.; Santhoshkumar, R.; Padmanabhan, B.; et al. Neuroanatomical zones of human traumatic brain injury reveal significant differences in protein profile and protein oxidation: Implications for secondary injury events. J. Neurochem. 2023, 167, 218–247. [Google Scholar] [CrossRef]
- Bielanin, J.P.; Metwally, S.A.H.; Paruchuri, S.S.; Sun, D. An overview of mild traumatic brain injuries and emerging therapeutic targets. Neurochem. Int. 2024, 172, 105655. [Google Scholar] [CrossRef]
- Kister, A.; Kister, I. Overview of myelin, major myelin lipids, and myelin-associated proteins. Front. Chem. 2023, 10, 1041961. [Google Scholar] [CrossRef]
- Svaren, J. MicroRNA and transcriptional crosstalk in myelinating glia. Neurochem. Int. 2014, 77, 50–57. [Google Scholar] [CrossRef]
- Wasik, N.; Sokol, B.; Holysz, M.; Manko, W.; Juszkat, R.; Jagodziński, P.P.; Jankowski, R. Serum myelin basic protein as a marker of brain injury in aneurysmal subarachnoid haemorrhage. Acta Neurochir. 2020, 162, 545–552. [Google Scholar] [CrossRef]
- Wu, S.L. Myelin basic protein in traumatic brain injury. In Biomarkers for Traumatic Brain Injury; Academic Press: Cambridge, MA, USA, 2020; pp. 221–224. [Google Scholar] [CrossRef]
- Ottens, A.K.; Golden, E.C.; Bustamante, L.; Hayes, R.L.; Denslow, N.D.; Wang, K.K. Proteolysis of multiple myelin basic protein isoforms after neurotrauma: Characterization by mass spectrometry. J. Neurochem. 2008, 104, 1404–1414. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, S.; Wirth, C.; Schmitz, W.; Trella, S.; Monoranu, C.M.; Ondruschka, B.; Bohnert, M. Myelin basic protein and neurofilament H in postmortem cerebrospinal fluid as surrogate markers of fatal traumatic brain injury. Int. J. Leg. Med. 2021, 135, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Su, E.; Bell, M.J.; Kochanek, P.M.; Wisniewski, S.R.; Bayır, H.; Clark, R.S.B.; Adelson, P.D.; Tyler-Kabara, E.C.; Janesko-Feldman, K.L.; Berger, R.P. Increased CSF concentrations of myelin basic protein after TBI in infants and children: Absence of significant effect of therapeutic hypothermia. Neurocritical Care 2012, 17, 401–407. [Google Scholar] [CrossRef]
- Berger, R.P.; Adelson, P.D.; Pierce, M.C.; Dulani, T.; Cassidy, L.D.; Kochanek, P.M. Serum neuron-specific enolase, S100B, and myelin basic protein concentrations after inflicted and noninflicted traumatic brain injury in children. J. Neurosurg. Pediatr. 2005, 103, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.P.; Adelson, P.D.; Richichi, R.; Kochanek, P.M. Serum biomarkers after traumatic and hypoxemic brain injuries: Insight into the biochemical response of the pediatric brain to inflicted brain injury. Dev. Neurosci. 2006, 28, 327–335. [Google Scholar] [CrossRef]
- Singh, A.; Singh, K.; Sahu, A.; Prasad, R.S.; Pandey, N.; Dhar, S. Serum Concentration of Myelin Basic Protein as a Prognostic Marker in Mild-to-moderate Head Injury Patients: A Prospective Study in a Tertiary Care Center. Indian J. Neurosurg. 2022, 11, 216–220. [Google Scholar] [CrossRef]
- Oehmichen, M.; Walter, T.; Meissner, C.; Friedrich, H. Time course of cortical hemorrhages after closed traumatic brain injury: Statistical analysis of posttraumatic histomorphological alterations. J. Neurotrauma 2003, 20, 87–103. [Google Scholar] [CrossRef]
- Businaro, R.; Fabrizi, C.; Caronti, B.; Calderaro, C.; Fumagalli, L.; Lauro, G.M. Myelin basic protein induces heme oxygenase-1 in human astroglial cells. Glia 2002, 37, 83–88. [Google Scholar] [CrossRef]
- Orr, M.E.; Sullivan, A.C.; Frost, B. A brief overview of tauopathy: Causes, consequences, and therapeutic strategies. Trend Pharmacol Sci. 2017, 38, 637–648. [Google Scholar] [CrossRef]
- Saha, P.; Sen, N. Tauopathy: A common mechanism for neurodegeneration and brain aging. Mech. Ageing Dev. 2019, 178, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Tsitsopoulos, P.P.; Marklund, N. Amyloid-β peptides and tau protein as biomarkers in cerebrospinal and interstitial fluid following traumatic brain injury: A review of experimental and clinical studies. Front. Neurol. 2013, 4, 79. [Google Scholar] [CrossRef] [PubMed]
- Edwards III, G.; Zhao, J.; Dash, P.K.; Soto, C.; Moreno-Gonzalez, I. Traumatic brain injury induces tau aggregation and spreading. J. Neurotrauma 2020, 37, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Alavi, N.; Seyedeh, M.; Soussi-Yanicostas, N. Tau Hyperphosphorylation and Oxidative Stress, a Critical Vicious Circle in Neurodegenerative Tauopathies? Oxidative Med. Cell. Longev. 2015, 2015, 151979. [Google Scholar] [CrossRef]
- Katsumoto, A.; Takeuchi, H.; Tanaka, F. Tau Pathology in Chronic Traumatic Encephalopathy and Alzheimer’s Disease: Similarities and Differences. Front. Neurol. 2019, 10, 980. [Google Scholar] [CrossRef]
- Diomede, L.; Zanier, E.R.; Moro, F.; Vegliante, G.; Colombo, L.; Russo, L.; Cagnotto, A.; Natale, C.; Xodo, F.M.; De Luigi, A.; et al. Aβ1-6A2V(D) peptide, effective on Aβ aggregation, inhibits tau misfolding and protects the brain after traumatic brain injury. Mol. Psychiatry 2023, 28, 2433–2444. [Google Scholar] [CrossRef]
- Rubenstein, R.; Chang, B.; Grinkina, N.; Drummond, E.; Davies, P.; Ruditzky, M.; Sharma, D.; Wang, K.; Wisniewski, T. Tau phosphorylation induced by severe closed head traumatic brain injury is linked to the cellular prion protein. Acta Neuropathol. Commun. 2017, 5, 30. [Google Scholar] [CrossRef]
- Liliang, P.C.; Liang, C.L.; Lu, K.; Wang, K.W.; Weng, H.C.; Hsieh, C.H.; Tsai, Y.; Chen, H.J. Relationship between injury severity and serum tau protein levels in traumatic brain injured rats. Resuscitation 2010, 81, 1205–1208. [Google Scholar] [CrossRef]
- Rizzi, L.; Grinberg, L.T. Exploring the significance of caspase-cleaved tau in tauopathies and as a complementary pathology to phospho-tau in Alzheimer’s disease: Implications for biomarker development and therapeutic targeting. Acta Neuropathol Commun. 2024, 12, 36. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Han, L.; Guo, S.; Wang, L.; Xiong, Z.; Chen, Z.; Chen, W.; Liang, J. Serum τ protein as a potential biomarker in the assessment of traumatic brain injury. Exp. Ther. Med. 2016, 11, 1147–1151. [Google Scholar] [CrossRef]
- Ni, P.; Qiao, Y.; Tong, W.; Zhao, C.; Zheng, P. Associations between serum tau, neurological outcome, and cognition following traumatic brain injury. Neurol. India 2020, 68, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Shahim, P.; Politis, A.; van der Merwe, A.; Moore, B.; Ekanayake, V.; Lippa, S.M.; Chou, Y.-Y.; Pham, D.L.; Butman, J.A.; Diaz-Arrastia, R.; et al. Time course and diagnostic utility of NfL, tau, GFAp, and UCH-L1 in subacute and chronic TBI. Neurology 2020, 95, e623–e636. [Google Scholar] [CrossRef]
- Bittar, A.; Bhatt, N.; Hasan, T.F.; Montalbano, M.; Puangmalai, N.; McAllen, S.; Ellsworth, A.; Murillo, M.C.; Taglialatela, G.; Lucke-Wold, B.; et al. Neurotoxic tau oligomers after single versus repetitive mild traumatic brain injury. Brain Commun. 2019, 1, fcz004. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, Q.; Zhang, Y.W.; Xu, H. Proteolytic processing of Alzheimer’s β-amyloid precursor protein. J. Neurochem. 2012, 120 (Suppl. S1), 9–21. [Google Scholar] [CrossRef] [PubMed]
- Baranello, R.J.; Bharani, K.L.; Padmaraju, V.; Chopra, N.; Lahiri, D.K.; Greig, N.H.; Pappolla, M.A.; Sambamurti, K. Amyloid-beta protein clearance and degradation (ABCD) pathways and their role in Alzheimer’s disease. Curr. Alzheimer Res. 2015, 12, 32–46. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Sivanandam, T.M.; Thakur, M.K. Traumatic brain injury: A risk factor for Alzheimer’s disease. Neurosci. Biobehav. Rev. 2012, 36, 1376–1381. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Traumatic brain injury and amyloid-β pathology: A link to Alzheimer’s disease? Nat. Rev. Neurosci. 2010, 11, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Raby, C.A.; Morganti-Kossmann, M.C.; Kossmann, T.; Stahel, P.F.; Watson, M.D.; Evans, L.M.; Mehta, P.D.; Spiegel, K.; Kuo, Y.; Roher, A.E.; et al. Traumatic Brain Injury Increases β-Amyloid Peptide 1-42 in Cerebrospinal Fluid. J. Neurochem. 2002, 71, 2505–2509. [Google Scholar] [CrossRef]
- Marklund, N.; Farrokhnia, N.; Hånell, A.; Vanmechelen, E.; Enblad, P.; Zetterberg, H.; Blennow, K.; Hillered, L. Monitoring of β-Amyloid Dynamics after Human Traumatic Brain Injury. J. Neurotrauma 2014, 31, 42–55. [Google Scholar] [CrossRef]
- Olsson, A.; Csajbok, L.; Ost, M.; Höglund, K.; Nylén, K.; Rosengren, L.; Nellgård, B.; Blennow, K. Marked increase of beta-amyloid(1-42) and amyloid precursor protein in ventricular cerebrospinal fluid after severe traumatic brain injury. J. Neurol. 2004, 251, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Padurariu, M.; Ciobica, A.; Lefter, R.; Serban, I.L.; Stefanescu, C.; Chirita, R. The oxidative stress hypothesis in Alzheimer’s disease. Psychiatr. Danub. 2013, 25, 401–409. [Google Scholar] [PubMed]
- Bird, S.M.; Sohrabi, H.R.; Sutton, T.A.; Weinborn, M.; Rainey-Smith, S.R.; Brown, B.; Patterson, L.; Taddei, K.; Gupta, V.; Carruthers, M.; et al. Cerebral amyloid-β accumulation and deposition following traumatic brain injury—A narrative review and meta-analysis of animal studies. Neurosci. Biobehav. Rev. 2016, 64, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Camilus, N.; Quintero Arias, C.; Martic, S. Role of Triggers on the Structural and Functional Facets of TAR DNA-binding Protein 43. Neuroscience 2023, 511, 110–130. [Google Scholar] [CrossRef]
- Özen, I.; Abu Hamdeh, S.; Ruscher, K.; Marklund, N. Traumatic brain injury causes early aggregation of beta-amyloid peptides and NOTCH3 reduction in vascular smooth muscle cells of leptomeningeal arteries. Acta Neuropathol. 2025, 149, 10. [Google Scholar] [CrossRef]
- Romero Tirado, M.A.; Pampin, B.; Gallego, G.; García-Caballero, L.; Varela Gómez, M. Beta-amiloid precursor protein (β-App) and diffuse axonal damage after head injuries: A forensic point of view. Med. Leg. Costa. Rica. 2022, 39, 37–50. [Google Scholar]
- Marklund, N.; Blennow, K.; Zetterberg, H.; Ronne-Engström, E.; Enblad, P.; Hillered, L. Monitoring of brain interstitial total tau and beta amyloid proteins by microdialysis in patients with traumatic brain injury. J. Neurosurg. 2009, 110, 1227–1237. [Google Scholar] [CrossRef]
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar] [CrossRef]
- Finsterer, J.; Burgunder, J.-M. Recent progress in the genetics of motor neuron disease. Eur. J. Med. Genet. 2014, 57, 103–112. [Google Scholar] [CrossRef]
- Warraich, S.T.; Yang, S.; Nicholson, G.A.; Blair, I.P. TDP-43: A DNA and RNA binding protein with roles in neurodegenerative diseases. Int. J. Biochem. Cell Biol. 2010, 42, 1606–1609. [Google Scholar] [CrossRef]
- Siuda, J.; Fujioka, S.; Wszolek, Z.K. Parkinsonian syndrome in familial frontotemporal dementia. Park. Relat. Disord. 2014, 20, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Chen, C. TDP-43 is a key molecule accelerating development of Alzheimer’s disease following traumatic brain injury. Neural Regen. Res. 2024, 19, 955–956. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Rademakers, R. The role of TDP-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr. Opin. Neurol. 2008, 21, 693. [Google Scholar] [CrossRef] [PubMed]
- Rajič Bumber, J.; Pilipović, K.; Janković, T.; Dolenec, P.; Gržeta, N.; Križ, J.; Župan, G. Repetitive traumatic brain injury is associated with TDP-43 alterations, neurodegeneration, and glial activation in mice. J. Neuropathol. Exp. Neurol. 2021, 80, 2–14. [Google Scholar] [CrossRef]
- Jo, M.; Lee, S.; Jeon, Y.-M.; Kim, S.; Kwon, Y.; Kim, H.-J. The role of TDP-43 propagation in neurodegenerative diseases: Integrating insights from clinical and experimental studies. Exp. Mol. Med. 2020, 52, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Garnier, C.; Devred, F.; Byrne, D.; Puppo, R.; Roman, A.Y.; Malesinski, S.; Golovin, A.V.; Lebrun, R.; Ninkina, N.N.; Tsvetkov, P.O. Zinc binding to RNA recognition motif of TDP-43 induces the formation of amyloid-like aggregates. Sci. Rep. 2017, 7, 6812. [Google Scholar] [CrossRef]
- Heyburn, L.; Sajja, V.S.; Long, J.B. The role of TDP-43 in military-relevant TBI and chronic neurodegeneration. Front. Neurol. 2019, 10, 680. [Google Scholar] [CrossRef]
- McKee, A.C.; Gavett, B.E.; Stern, R.A.; Nowinski, C.J.; Cantu, R.C.; Kowall, N.W.; Perl, D.P.; Hedley-Whyte, E.T.; Price, B.; Sullivan, C.; et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J. Neuropathol. Exp. Neurol. 2010, 69, 918–929. [Google Scholar] [CrossRef]
- Janković, T.; Dolenec, P.; Rajič Bumber, J.; Gržeta, N.; Kriz, J.; Župan, G.; Pilipović, K. Differential Expression Patterns of TDP-43 in Single Moderate versus Repetitive Mild Traumatic Brain Injury in Mice. Int. J. Mol. Sci. 2021, 22, 12211. [Google Scholar] [CrossRef]
- Tremblay, C.; St-Amour, I.; Schneider, J.; Bennett, D.A.; Calon, F. Accumulation of transactive response DNA binding protein 43 in mild cognitive impairment and Alzheimer disease. J. Neuropathol. Exp. Neurol. 2011, 70, 788–798. [Google Scholar] [CrossRef]
- Bjorklund, G.R.; Wong, J.; Brafman, D.; Bowser, R.; Stabenfeldt, S.E. Traumatic brain injury induces TDP-43 mislocalization and neurodegenerative effects in tissue distal to the primary injury site in a non-transgenic mouse. Acta Neuropathol. Commun. 2023, 11, 137. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Hu, M.; Zhang, J.; Hashem, J.; Chen, C. TDP-43 drives synaptic and cognitive deterioration following traumatic brain injury. Acta Neuropathol. 2022, 144, 187–210. [Google Scholar] [CrossRef] [PubMed]
- Jamerlan, A.; An, S.S.A. The influence of Aβ-dependent and independent pathways on TDP-43 proteinopathy in Alzheimer’s disease: A possible connection to LATE-NC. Neurobiol. Aging. 2020, 95, 161–167. [Google Scholar] [CrossRef]
- Martin, E.J.; Santacruz, C.; Mitevska, A.; Jones, I.E.; Krishnan, G.; Gao, F.B.; Finan, J.D.; Kiskinis, E. Traumatic injury causes selective degeneration and TDP-43 mislocalization in human iPSC-derived C9orf72-associated ALS/FTD motor neurons. bioRxiv 2024. [Google Scholar] [CrossRef]
- Nag, S.; Schneider, J.A. Limbic-predominant age-related TDP43 encephalopathy (LATE) neuropathological change in neurodegenerative diseases. Nat. Rev. Neurol. 2023, 19, 525–541. [Google Scholar] [CrossRef]
- Pagnotti, R.M.B.; Pudumjee, S.B.; Cross, C.L.; Miller, J.B. Cognitive and clinical characteristics of patients with limbic-predominant age-related TDP-43 encephalopathy. Neurology 2023, 100, e2027-35. [Google Scholar]
- Anderson, E.N.; A Morera, A.; Kour, S.; Cherry, J.D.; Ramesh, N.; Gleixner, A.; Schwartz, J.C.; Ebmeier, C.; Old, W.; Donnelly, C.J.; et al. Traumatic injury compromises nucleocytoplasmic transport and leads to TDP-43 pathology. eLife 2021, 10, e67587. [Google Scholar] [CrossRef]
- Colombrita, C.; Zennaro, E.; Fallini, C.; Weber, M.; Sommacal, A.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 2009, 111, 1051–1061. [Google Scholar] [CrossRef]
- Pisciottani, A.; Croci, L.; Lauria, F.; Marullo, C.; Savino, E.; Ambrosi, A.; Podini, P.; Marchioretto, M.; Casoni, F.; Cremona, O.; et al. Neuronal models of TDP-43 proteinopathy display reduced axonal translation, increased oxidative stress, and defective exocytosis. Front. Cell. Neurosci. 2023, 17, 1253543. [Google Scholar] [CrossRef]
- Sessa, F.; Maglietta, F.; Bertozzi, G.; Salerno, M.; Di Mizio, G.; Messina, G.; Montana, A.; Ricci, P.; Pomara, C. Human Brain Injury and miRNAs: An Experimental Study. Int. J. Mol. Sci. 2019, 20, 1546. [Google Scholar] [CrossRef]
- Valinezhad Orang, A.; Safaralizadeh, R.; Kazemzadeh-Bavili, M. Mechanisms of miRNA-Mediated Gene Regulation from Common Downregulation to mRNA-Specific Upregulation. Int. J. Genomics 2014, 2014, 970607. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Atif, H.; Hicks, S.D. A Review of MicroRNA Biomarkers in Traumatic Brain Injury. J. Exp. Neurosci. 2019, 13, 1179069519832286. [Google Scholar] [CrossRef] [PubMed]
- Pinchi, E.; Frati, P.; Arcangeli, M.; Volonnino, G.; Tomassi, R.; Santoro, P.; Cipolloni, L. MicroRNAs: The New Challenge for Traumatic Brain Injury Diagnosis. Curr. Neuropharmacol. 2020, 18, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Consalvo, F.; Padovano, M.; Scopetti, M.; Morena, D.; Cipolloni, L.; Fineschi, V.; Santurro, A. Analysis of miRNA Expression Profiles in Traumatic Brain Injury (TBI) and Their Correlation with Survival and Severity of Injury. Int. J. Mol. Sci. 2024, 25, 9539. [Google Scholar] [CrossRef]
- Mavroudis, I.; Petridis, F.; Balmus, I.-M.; Ciobica, A.; Gorgan, D.L.; Luca, A.C. Review on the Role of Salivary Biomarkers in the Diagnosis of Mild Traumatic Brain Injury and Post-Concussion Syndrome. Diagnostics 2023, 13, 1367. [Google Scholar] [CrossRef]
- Di Pietro, V.; Porto, E.; Ragusa, M.; Barbagallo, C.; Davies, D.; Forcione, M.; Logan, A.; Di Pietro, C.; Purrello, M.; Grey, M.; et al. Salivary MicroRNAs: Diagnostic Markers of Mild Traumatic Brain Injury in Contact-Sport. Front. Mol. Neurosci. 2018, 11, 290. [Google Scholar] [CrossRef]
- Di Pietro, V.; Ragusa, M.; Davies, D.; Su, Z.; Hazeldine, J.; Lazzarino, G.; Hill, L.J.; Crombie, N.; Foster, M.; Purrello, M.; et al. MicroRNAs as Novel Biomarkers for the Diagnosis and Prognosis of Mild and Severe Traumatic Brain Injury. J. Neurotrauma 2017, 34, 1948–1956. [Google Scholar] [CrossRef]
- Liu, L.; Sun, T.; Liu, Z.; Chen, X.; Zhao, L.; Qu, G.; Li, Q. Traumatic brain injury dysregulates microRNAs to modulate cell signaling in rat hippocampus. PLoS ONE 2014, 9, e103948. [Google Scholar] [CrossRef]
- Redell, J.B.; Zhao, J.; Dash, P.K. Altered expression of miRNA-21 and its targets in the hippocampus after traumatic brain injury. J. Neurosci. Res. 2011, 89, 212–221. [Google Scholar] [CrossRef]
- Weisz, H.A.; Kennedy, D.; Widen, S.; Spratt, H.; Sell, S.L.; Bailey, C.; Sheffield-Moore, M.; DeWitt, D.S.; Prough, D.S.; Levin, H.; et al. MicroRNA sequencing of rat hippocampus and human biofluids identifies acute, chronic, focal and diffuse traumatic brain injuries. Sci. Rep. 2020, 10, 3341. [Google Scholar] [CrossRef] [PubMed]
- Mitra, B.; Major, B.P.; Reyes, J.; Surendran, N.; Bain, J.; Giesler, L.P.; O’Brien, W.T.; Sorich, E.; Willmott, C.; Shultz, S.R.; et al. MicroRNA biomarkers for diagnosis of mild traumatic brain injury and prediction of persistent symptoms: A prospective cohort study. J. Clin. Neurosci. 2023, 115, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Sessa, F.; Pomara, C.; Schembari, F.; Esposito, M.; Capasso, E.; Pesaresi, M.; Osuna, E.; Ulas, E.; Zammit, C.; Salerno, M. MiRNA Dysregulation in Brain Injury: An In Silico Study to Clarify the Role of a MiRNA Set. Curr. Neuropharmacol. 2025, 23, 209–231. [Google Scholar] [CrossRef]
- Musso, N.; Bivona, D.; Bonomo, C.; Bonacci, P.; D’ippolito, M.E.; Boccagni, C.; Rubino, F.; De Tanti, A.; Lucca, L.F.; Pingue, V.; et al. Investigating microRNAs as biomarkers in disorders of consciousness: A longitudinal multicenter study. Sci. Rep. 2023, 13, 18415. [Google Scholar] [CrossRef]
- Yang, J.; Han, H.; Zhao, Y.; Qin, H. Specific miRNA and its target in neutrophils after traumatic injury. Acta Biochim. Et Biophys. Sin. 2015, 47, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.; Gao, M.; Xu, W.; Sun, X.; Chen, T.; Xu, H.; Qiu, H. Mechanisms of microRNA-132 in central neurodegenerative diseases: A comprehensive review. Biomed. Pharmacother. 2024, 170, 116029. [Google Scholar] [CrossRef]
- Jiang, H.; Liu, J.; Guo, S.; Zeng, L.; Cai, Z.; Zhang, J.; Wang, L.; Li, Z.; Liu, R. miR-23b-3p rescues cognition in Alzheimer’s disease by reducing tau phosphorylation and apoptosis via GSK-3β signaling pathways. Mol. Ther. Nucleic Acids 2022, 28, 539–557. [Google Scholar] [CrossRef]
- Li, Z.; Xu, R.; Zhu, X.; Li, Y.; Wang, Y.; Xu, W. MicroRNA-23a-3p improves traumatic brain injury through modulating the neurological apoptosis and inflammation response in mice. Cell Cycle 2020, 19, 24–38. [Google Scholar] [CrossRef]
- Sun, P.; Liu, D.Z.; Jickling, G.C.; Sharp, F.R.; Yin, K.J. MicroRNA-based therapeutics in central nervous system injuries. J. Cereb. Blood Flow Metab. 2018, 38, 1125–1148. [Google Scholar] [CrossRef]
- Albano, G.D.; Stassi, C.; Argo, A.; Zerbo, S. An Overview on the Use of miRNAs as Possible Forensic Biomarkers for the Diagnosis of Traumatic Brain Injury. Int. J. Mol. Sci. 2023, 24, 6503. [Google Scholar] [CrossRef]
- Sawant, H.; Sun, B.; Mcgrady, E.; Bihl, J.C. Role of miRNAs in neurovascular injury and repair. J. Cereb. Blood Flow Metab. 2024, 44, 1693–1708. [Google Scholar] [CrossRef] [PubMed]
- Konovalova, J.; Gerasymchuk, D.; Parkkinen, I.; Chmielarz, P.; Domanskyi, A. Interplay between MicroRNAs and Oxidative Stress in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 6055. [Google Scholar] [CrossRef]
- Wang, W.X.; Visavadiya, N.P.; Pandya, J.D.; Nelson, P.T.; Sullivan, P.G.; Springer, J.E. Mitochondria-associated microRNAs in rat hippocampus following traumatic brain injury. Exp. Neurol. 2015, 265, 84–93. [Google Scholar] [CrossRef]
- Zhu, Z.; Huang, X.; Du, M.; Wu, C.; Fu, J.; Tan, W.; Wu, B.; Zhang, J.; Liao, Z.B. Recent advances in the role of miRNAs in post-traumatic stress disorder and traumatic brain injury. Mol. Psychiatry 2023, 28, 2630–2644. [Google Scholar] [CrossRef]
- Saha, S. Role of microRNA in Oxidative Stress. Stresses 2024, 4, 269–281. [Google Scholar] [CrossRef]
- Xu, W.; Li, F.; Liu, Z.; Xu, Z.; Sun, B.; Cao, J.; Liu, Y. MicroRNA-27b inhibition promotes Nrf2/ARE pathway activation and alleviates intracerebral hemorrhage-induced brain injury. Oncotarget 2017, 8, 70669–70684. [Google Scholar] [CrossRef]
- Wu, P.; He, B.; Li, X.; Zhang, H. Roles of microRNA-124 in traumatic brain injury: A comprehensive review. Front. Cell. Neurosci. 2023, 17, 1298508. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Han, K.; Zhang, D.; Chen, J.; Xu, Z.; Hou, L. The role of long noncoding RNA in traumatic brain injury. Neuropsychiatr. Dis. Treat. 2019, 15, 1671–1677. [Google Scholar] [CrossRef]
- Roberts, T.C.; Morris, K.V.; Wood, M.J. The role of long non-coding RNAs in neurodevelopment, brain function and neurological disease. Philos. Trans. R. Soc. B. Biol. Sci. 2014, 369, 20130507. [Google Scholar] [CrossRef]
- Liu, S.J.; Nowakowski, T.J.; Pollen, A.A.; Lui, J.H.; Horlbeck, M.A.; Attenello, F.J.; He, D.; Weissman, J.S.; Kriegstein, A.R.; Diaz, A.A.; et al. Single-cell analysis of long non-coding RNAs in the developing human neocortex. Genome Biol. 2016, 17, 67. [Google Scholar] [CrossRef] [PubMed]
- Navandar, M.; Vennin, C.; Lutz, B.; Gerber, S. Long non-coding RNAs expression and regulation across different brain regions in primates. Sci. Data 2024, 11, 545. [Google Scholar] [CrossRef]
- Alammari, F.; Al-Hujaily, E.M.; Alshareeda, A.; Albarakati, N.; Al-Sowayan, B.S. Hidden regulators: The emerging roles of lncRNAs in brain development and disease. Front. Neurosci. 2024, 18, 1392688. [Google Scholar] [CrossRef]
- Yu, Y.; Cao, F.; Ran, Q.; Wang, F. Long non-coding RNA Gm4419 promotes trauma-induced astrocyte apoptosis by targeting tumor necrosis factor α. Biochem. Biophys. Res. Commun. 2017, 491, 478–485. [Google Scholar] [CrossRef]
- Lim, K.H.; Yang, S.; Kim, S.H.; Chun, S.; Joo, J.Y. Discoveries for Long Non-Coding RNA Dynamics in Traumatic Brain Injury. Biology 2020, 9, 458. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, X.; Luo, W.; Wu, Y.; Tang, T.; Wang, Y. The Involvement of Long Non-coding RNA and Messenger RNA Based Molecular Networks and Pathways in the Subacute Phase of Traumatic Brain Injury in Adult Mice. Front. Neuroinformatics 2022, 16, 794342. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Chen, W.; Cao, K.; Wang, Z.; Zheng, P. Expression Profiles of Long Non-coding RNA and Messenger RNA in Human Traumatic Brain Injury. Mol. Ther. Nucleic Acids 2020, 22, 99–113. [Google Scholar] [CrossRef]
- Lei, J.; Zhang, X.; Tan, R.; Li, Y.; Zhao, K.; Niu, H. Levels of lncRNA GAS5 in Plasma of Patients with Severe Traumatic Brain Injury: Correlation with Systemic Inflammation and Early Outcome. J. Clin. Med. 2022, 11, 3319. [Google Scholar] [CrossRef]
- Tang, Z.; Xu, S.; Zhao, S.; Luo, Z.; Tang, Y.; Zhang, Y. Clinical value of serum LncRNA MIAT in early diagnosis and prognosis assessment of traumatic brain injury. Clin. Neurol. Neurosurg. 2024, 249, 108648. [Google Scholar] [CrossRef]
- Rabou, Y.K.A.; Zayed, A.A.; Fahim, S.A.; Abdelgwad, M.; El Fiki, A.; Fayed, N.N. Exploring New and Promising Genetic Biomarkers for Evaluating Traumatic Brain Injuries: A Case-Control Study. Neurochem. Res. 2025, 50, 48. [Google Scholar] [CrossRef]
- Patel, R.S.; Krause-Hauch, M.; Kenney, K.; Miles, S.; Nakase-Richardson, R.; Patel, N.A. Long Noncoding RNA VLDLR-AS1 Levels in Serum Correlate with Combat-Related Chronic Mild Traumatic Brain Injury and Depression Symptoms in US Veterans. Int. J. Mol. Sci. 2024, 25, 1473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, H. Long Non-coding RNA in CNS Injuries: A New Target for Therapeutic Intervention. Mol. Ther. Nucleic Acids 2019, 17, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, Y. Regulation of oxidative stress by long non-coding RNAs in central nervous system disorders. Front. Mol. Neurosci. 2022, 15, 931704. [Google Scholar] [CrossRef]
- Ryan, A.K.; Rich, W.; Reilly, M.A. Oxidative stress in the brain and retina after traumatic injury. Front. Neurosci. 2023, 17, 1021152. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, C.; Crupi, R.; Calabrese, V.; Graziano, A.; Milone, P.; Pennisi, G.; Radak, Z.; Calabrese, E.J.; Cuzzocrea, S. Traumatic brain injury: Oxidative stress and neuroprotection. Antioxid. Redox signaling 2013, 19, 836–853. [Google Scholar] [CrossRef]
- Wu, X.; Wei, H.; Wu, J.Q. Coding and long non-coding gene expression changes in the CNS traumatic injuries. Cell. Mol. Life Sci. 2022, 79, 123. [Google Scholar] [CrossRef]
- Wang, J.P.; Li, C.; Ding, W.C.; Peng, G.; Xiao, G.L.; Chen, R.; Cheng, Q. Research Progress on the Inflammatory Effects of Long Non-coding RNA in Traumatic Brain Injury. Front. Mol. Neurosci. 2022, 15, 835012. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Hamblin, M.H.; Yin, K.J. Non-coding RNAs in the regulation of blood-brain barrier functions in central nervous system disorders. Fluids Barriers CNS 2022, 19, 27. [Google Scholar] [CrossRef]
- Qiao, L.X.; Zhao, R.B.; Wu, M.F.; Zhu, L.H.; Xia, Z.K. Silencing of long non-coding antisense RNA brain-derived neurotrophic factor attenuates hypoxia/ischemia-induced neonatal brain injury. Int. J. Mol. Med. 2020, 46, 653–662. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ionescu, C.; Ghidersa, M.; Ciobica, A.; Mavroudis, I.; Kazis, D.; Petridis, F.E.; Gorgan, D.L.; Balmus, I.-M. Potential Correlation Between Molecular Biomarkers and Oxidative Stress in Traumatic Brain Injury. Int. J. Mol. Sci. 2025, 26, 3858. https://doi.org/10.3390/ijms26083858
Ionescu C, Ghidersa M, Ciobica A, Mavroudis I, Kazis D, Petridis FE, Gorgan DL, Balmus I-M. Potential Correlation Between Molecular Biomarkers and Oxidative Stress in Traumatic Brain Injury. International Journal of Molecular Sciences. 2025; 26(8):3858. https://doi.org/10.3390/ijms26083858
Chicago/Turabian StyleIonescu, Cătălina, Madalina Ghidersa, Alin Ciobica, Ioannis Mavroudis, Dimitrios Kazis, Foivos E. Petridis, Dragoș Lucian Gorgan, and Ioana-Miruna Balmus. 2025. "Potential Correlation Between Molecular Biomarkers and Oxidative Stress in Traumatic Brain Injury" International Journal of Molecular Sciences 26, no. 8: 3858. https://doi.org/10.3390/ijms26083858
APA StyleIonescu, C., Ghidersa, M., Ciobica, A., Mavroudis, I., Kazis, D., Petridis, F. E., Gorgan, D. L., & Balmus, I.-M. (2025). Potential Correlation Between Molecular Biomarkers and Oxidative Stress in Traumatic Brain Injury. International Journal of Molecular Sciences, 26(8), 3858. https://doi.org/10.3390/ijms26083858