
Myristoyl-CM4 Exhibits Direct Anticancer Activity and Immune Modulation in Hepatocellular Carcinoma: Evidence from In Vitro and Mouse Model Studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
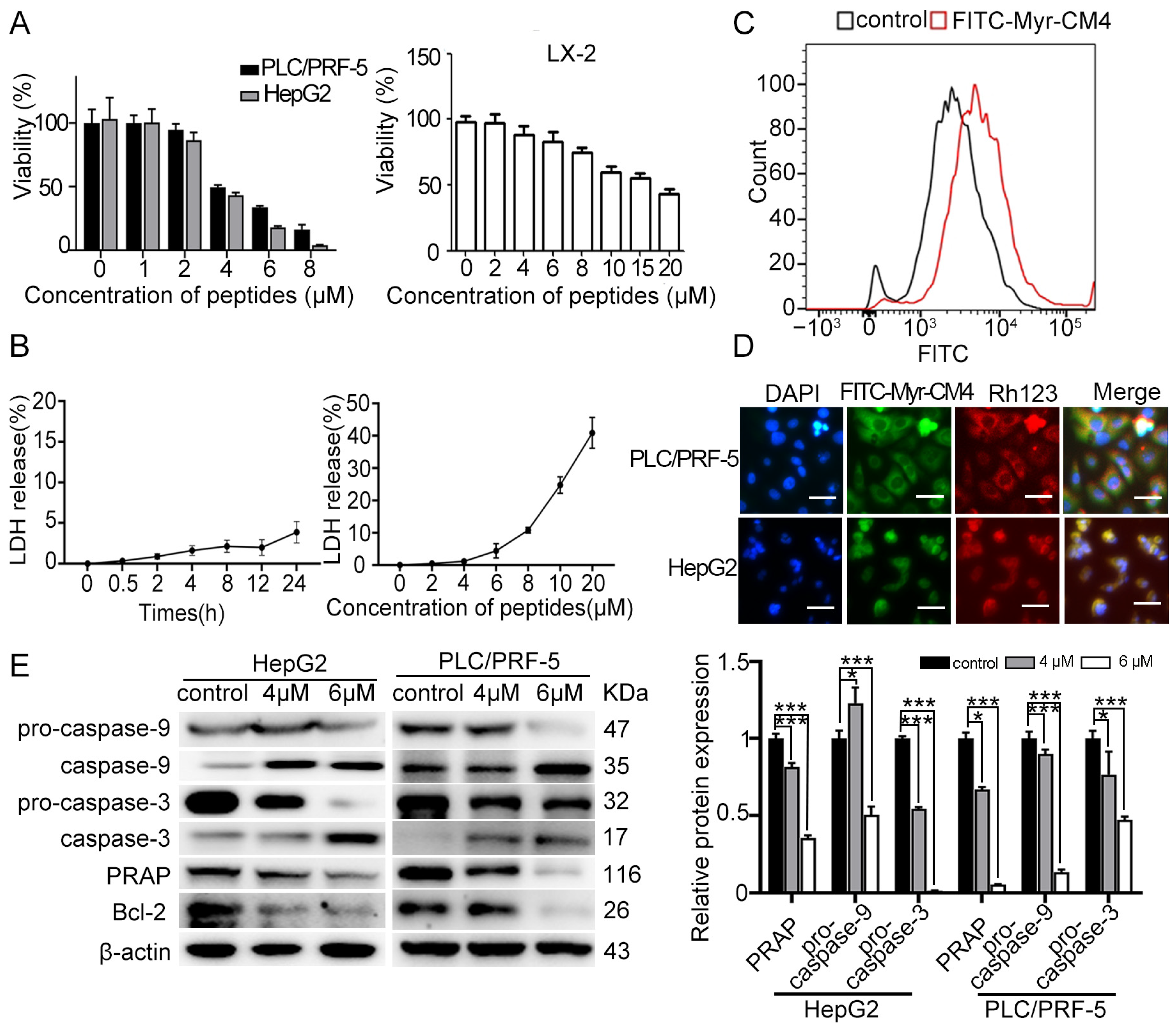
2.1. Myristoyl-CM4 Induces Cytotoxicity in HCC Cells
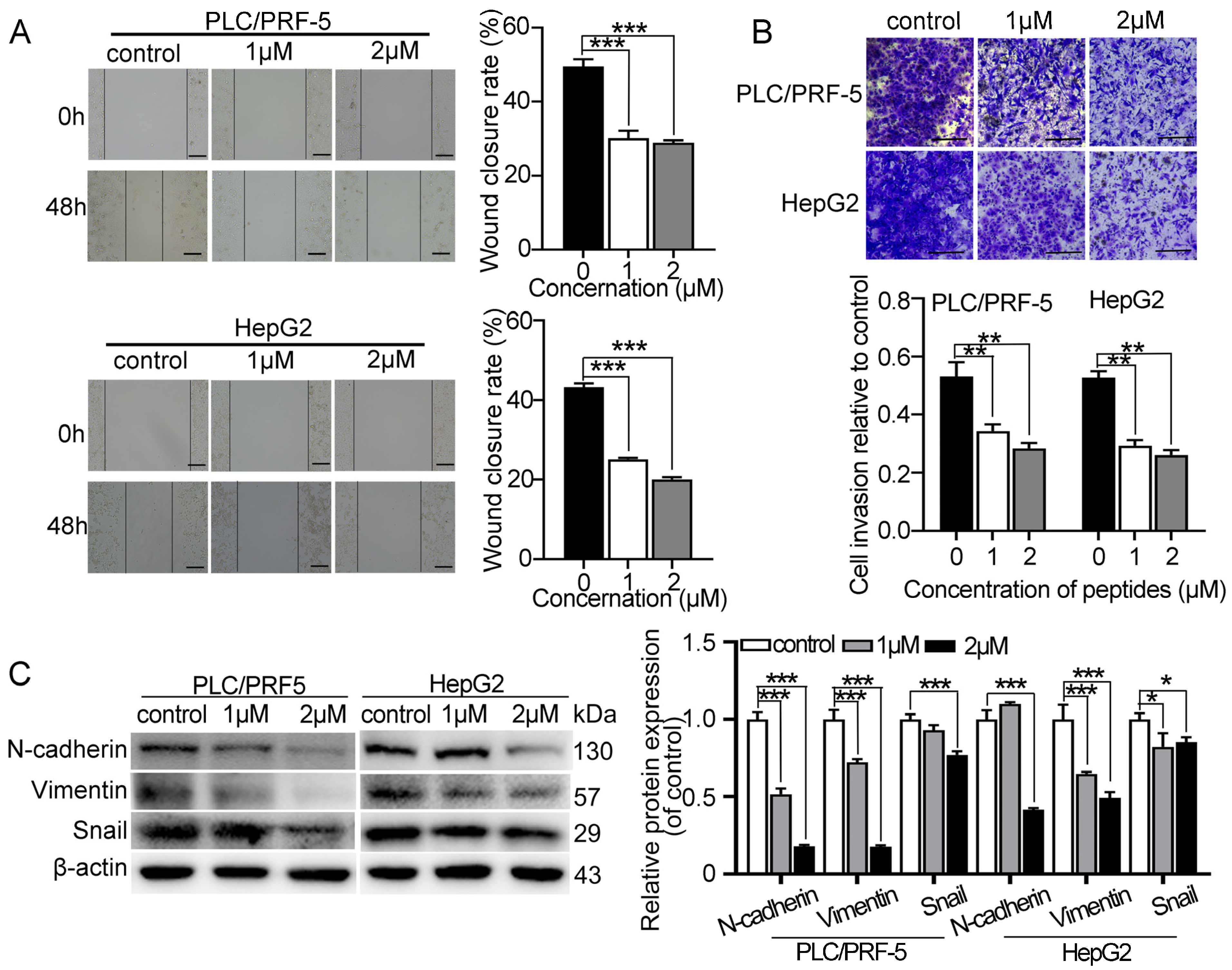
2.2. Myristoyl-CM4 Inhibits Migration and Invasion of Cultured HCC Cells
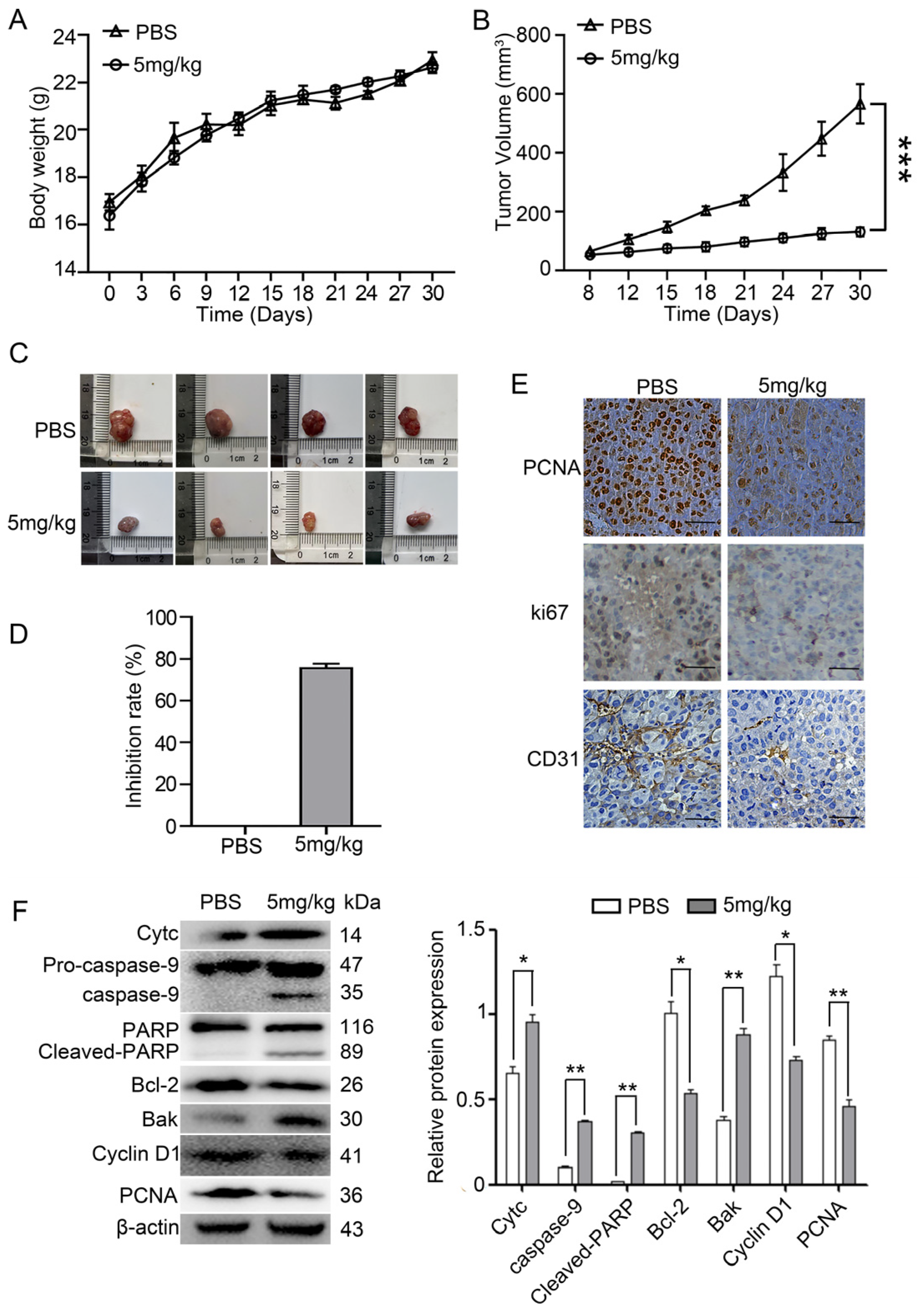
2.3. Myristoyl-CM4 Inhibits the Growth of PLC/PRF-5 Xenograft Tumor Growth
2.4. Myristoyl-CM4 Inhibits HCC Cell Survival in the HCC/Macrophage Co-Culture System
2.5. Myristoyl-CM4 Inhibits Proliferation, EMT, Migration, and Invasion of HCC Cells in the HCC/M0 Co-Culture System
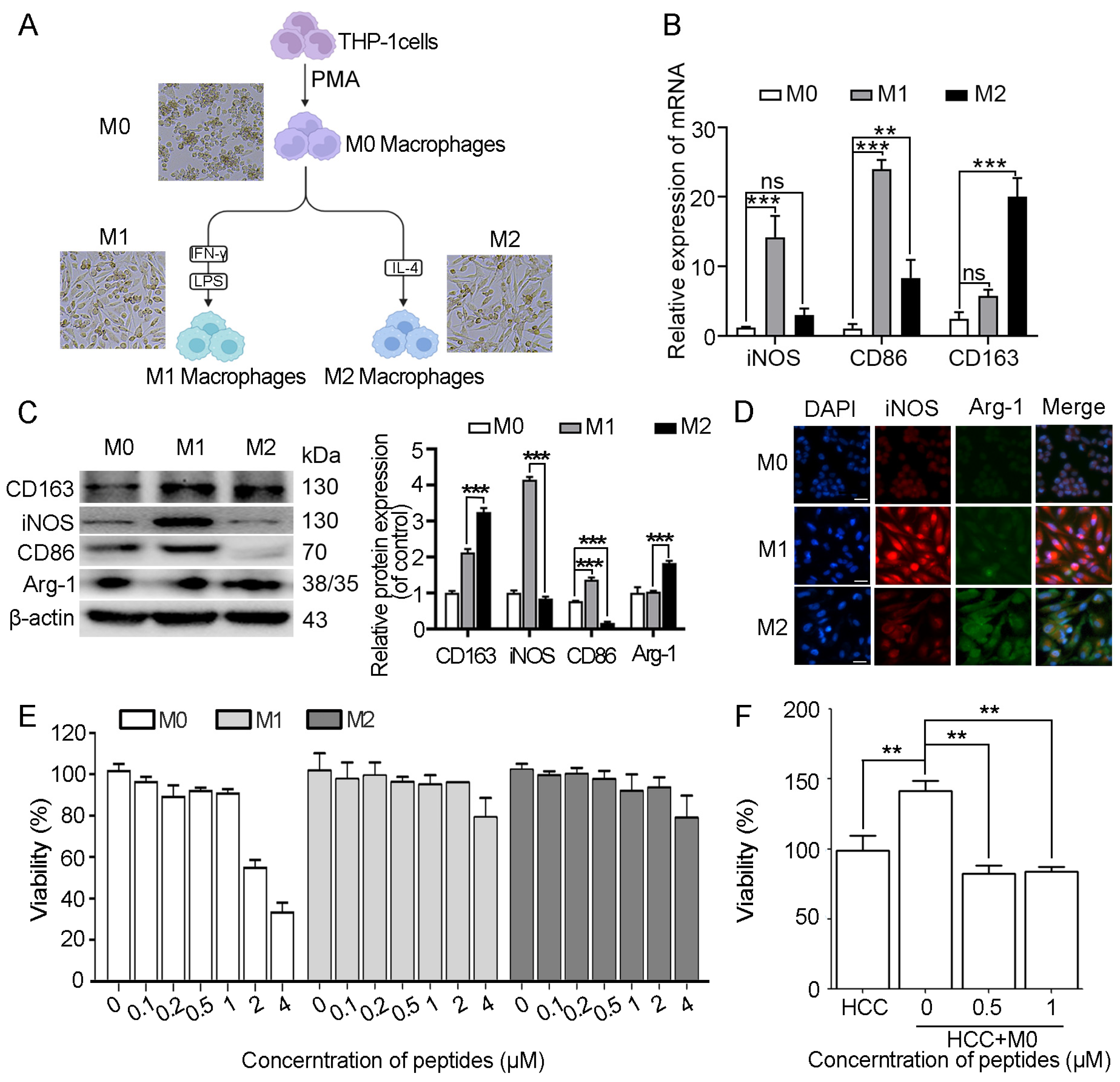
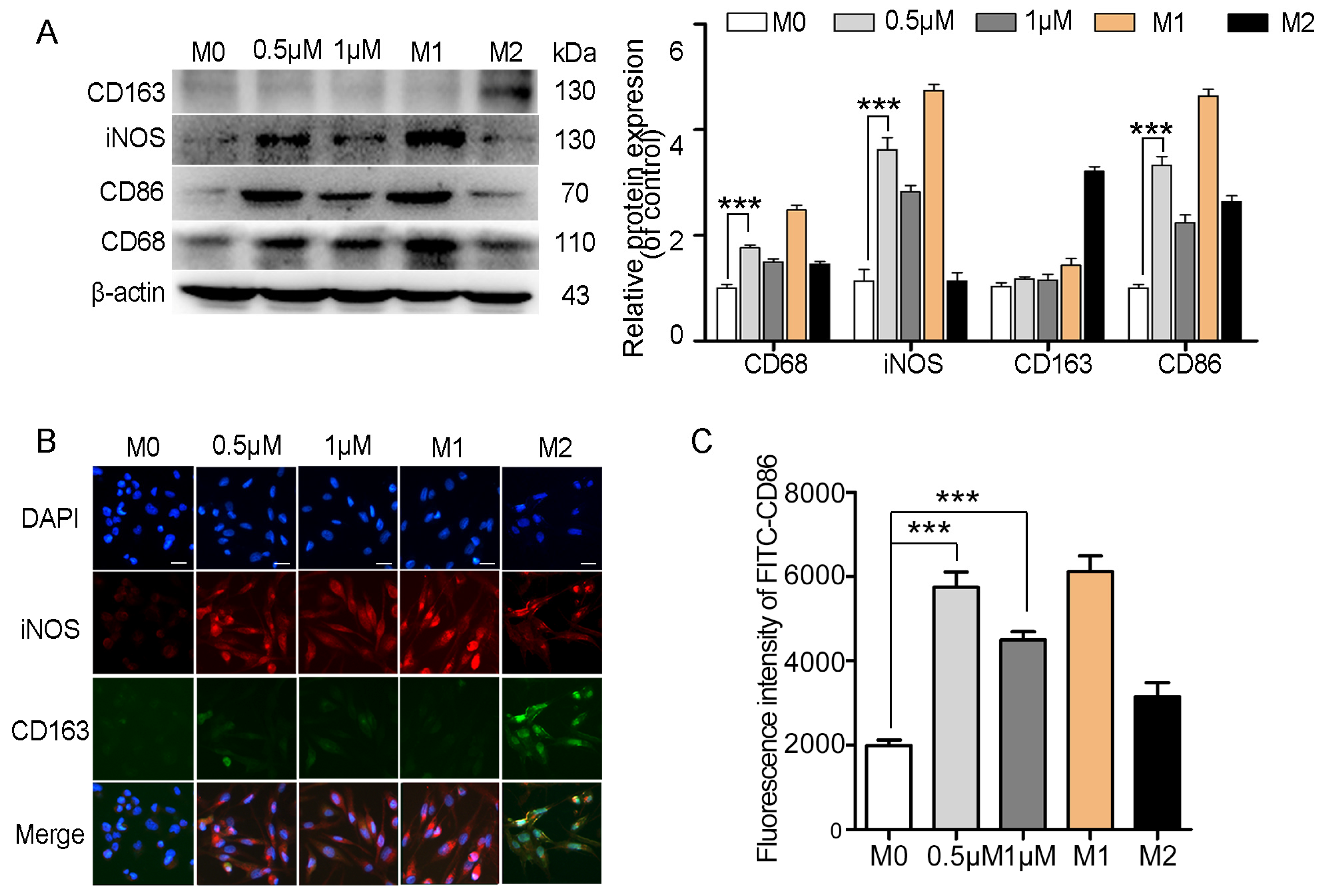
2.6. Myristoyl-CM4 Promotes Macrophage M1 Polarization
2.7. Myristoyl-CM4 Regulates Macrophage Polarization and Inhibits Tumor Growth in Co-Xenografts Tumor Models
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Macrophage Differentiation and Polarization
4.4. CCK-8 Assay
4.5. MTT Assay
4.6. Lactate Dehydrogenase (LDH) Release Assay
4.7. Cell Binding Assay and Rh123 Staining
4.8. Western Blotting
4.9. Quantitative Real-Time PCR (qRT-PCR)
4.10. Wound Healing Assay
4.11. Transwell Invasion Assay
4.12. EdU Incorporation Assay
4.13. Immunofluorescent Staining
4.14. Xenograft Tumor Model in Nude Mice
4.15. Immunohistochemistry (IHC)
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Cossiga, V.; Guarino, M.; Ranieri, L.; Morisco, F. The Role of Hepatitis Viruses as Drivers of Hepatocancerogenesis. Cancers 2024, 16, 1505. [Google Scholar] [CrossRef] [PubMed]
- Becht, R.; Kiełbowski, K.; Wasilewicz, M.P. New Opportunities in the Systemic Treatment of Hepatocellular Carcinoma-Today and Tomorrow. Int. J. Mol. Sci. 2024, 25, 1456. [Google Scholar] [CrossRef] [PubMed]
- Ladd, A.D.; Duarte, S.; Sahin, I.; Zarrinpar, A. Mechanisms of drug resistance in HCC. Hepatology 2024, 79, 926–940. [Google Scholar] [CrossRef]
- Safri, F.; Nguyen, R.; Zerehpooshnesfchi, S.; George, J.; Qiao, L. Heterogeneity of hepatocellular carcinoma: From mechanisms to clinical implications. Cancer Gene Ther. 2024, 31, 1105–1112. [Google Scholar] [CrossRef]
- Oura, K.; Morishita, A.; Tani, J.; Masaki, T. Tumor immune microenvironment and immunosuppressive therapy in hepatocellular carcinoma: A review. Int. J. Mol. Sci. 2021, 22, 5801. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, W.; Li, Q. Potential therapeutic targets in the tumor microenvironment of hepatocellular carcinoma: Reversing the protumor effect of tumor-associated macrophages. J. Exp. Clin. Cancer. Res. 2021, 40, 73. [Google Scholar] [CrossRef]
- Soon, T.N.; Chia, A.Y.Y.; Yap, W.H.; Tang, Y.Q. Anticancer mechanisms of bioactive peptides. Protein Pept. Lett. 2020, 27, 823–830. [Google Scholar] [CrossRef]
- Lee, C.; Bae, S.S.; Joo, H.; Bae, H. Melittin suppresses tumor progression by regulating tumor-associated macrophages in a Lewis lung carcinoma mouse model. Oncotarget 2017, 8, 54951–54965. [Google Scholar] [CrossRef]
- Han, I.H.; Jeong, C.; Yang, J.; Park, S.H.; Hwang, D.S.; Bae, H. Therapeutic Effect of Melittin-dKLA Targeting Tumor-Associated Macrophages in Melanoma. Int. J. Mol. Sci. 2022, 23, 3094. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Choi, I.; Han, I.H.; Bae, H. Enhanced Therapeutic Effect of Optimized Melittin-dKLA, a Peptide Agent Targeting M2-like Tumor-Associated Macrophages in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2022, 23, 15751. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, L.; He, D.; Wu, Y.; Liu, X.; Yang, Y.; Chen, Z.; Dong, Z.; Luo, Y.; Song, Y. Application value of antimicrobial peptides in gastrointestinal tumors. Int. J. Mol. Sci. 2023, 24, 16718. [Google Scholar] [CrossRef]
- Wu, J.; Deng, R.; Yan, J.; Zhu, B.; Wang, J.; Xu, Y.; Gui, S.; Jin, X.; Lu, X. A cell transmembrane peptide chimeric M(27–39)-HTPP targeted therapy for hepatocellular carcinoma. iScience 2023, 26, 106766. [Google Scholar] [CrossRef]
- Zeng, J.; Wang, J.; Wu, J.; Deng, R.; Zhang, L.; Chen, Q.; Wang, J.; Jin, X.; Gui, S.; Xu, Y.; et al. A novel antimicrobial peptide M1–8 targets the lysosomal pathway to inhibit autolysosome formation and promote apoptosis in liver cancer cells. J. Cell Mol. Med. 2023, 27, 340–352. [Google Scholar] [CrossRef]
- Li, X.; Song, W.; Zhang, M.; Zhao, P. Human β-defensin 1 Functions as a Tumor Suppressor via ER Stress-triggered JNK pathway in Hepatocellular Carcinoma. J. BUON 2021, 26, 1365–1372. [Google Scholar]
- Yang, Y.Q.; Zhang, H.D.; Wanyan, Y.K.; Liu, K.H.; Lv, T.T.; Li, M.; Chen, Y.Q. Effect of hydrophobicity on the anticancer activity of fatty-acyl-conjugated CM4 in breast cancer cells. ACS Omega 2020, 5, 21513–21523. [Google Scholar] [CrossRef]
- Li, C.; Liu, H.; Yang, Y.; Xu, X.; Lv, T.; Zhang, H.; Liu, K.; Zhang, S.; Chen, Y. N-myristoylation of antimicrobial peptide CM4 enhances its anticancer activity by interacting with cell membrane and targeting mitochondria in breast cancer cells. Front. Pharmacol. 2018, 9, 1297. [Google Scholar] [CrossRef]
- Zhang, H.; Han, D.; Lv, T.; Liu, K.; Yang, Y.; Xu, X.; Chen, Y. Novel peptide myristoly-CM4 induces selective cytotoxicity in leukemia K562/MDR and Jurkat cells by necrosis and/or apoptosis pathway. Drug Des. Devel. Ther. 2019, 13, 2153–2167. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, C.; Zhang, W.; Li, X. Bioactive peptides for anticancer therapies. Biomater. Transl. 2023, 4, 5–17. [Google Scholar]
- Stela, M.; Cichon, N.; Spławska, A.; Szyposzynska, M.; Bijak, M. Therapeutic Potential and Mechanisms of Bee Venom Therapy: A Comprehensive Review of Apitoxin Applications and Safety Enhancement Strategies. Pharmaceuticals 2024, 17, 1211. [Google Scholar] [CrossRef] [PubMed]
- Min, K.A.; Maharjan, P.; Ham, S.; Shin, M.C. Pro-apoptotic peptides-based cancer therapies: Challenges and strategies to enhance therapeutic efficacy. Arch. Pharm. Res. 2018, 41, 594–616. [Google Scholar] [CrossRef] [PubMed]
- Chiangjong, W.; Chutipngtanate, S.; Hongeng, S. Anticancer peptide: Physicochemical property, functional aspect and trend in clinical application (Review). Int. J. Oncol. 2020, 57, 678–696. [Google Scholar] [CrossRef]
- Sas, Z.; Cendrowicz, E.; Weinhäuser, I.; Rygiel, T.P. Tumor microenvironment of hepatocellular carcinoma: Challenges and opportunities for new treatment options. Int. J. Mol. Sci. 2022, 23, 3778. [Google Scholar] [CrossRef]
- Uchino, K.; Tateishi, R.; Shiina, S.; Kanda, M.; Masuzaki, R.; Kondo, Y.; Goto, T.; Omata, M.; Yoshida, H.; Koike, K. Hepatocellular carcinoma with extrahepatic metastasis: Clinical features and prognostic factors. Cancer 2011, 117, 4475–4483. [Google Scholar] [CrossRef]
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, plasticity, and tumor metastasis. Trends Cell Biol. 2020, 30, 764–776. [Google Scholar] [CrossRef]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-cadherin and N-cadherin switch in epithelial-to-mesenchymal transition: Signaling, therapeutic implications, and challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Haque, S.; Hussain, A.; Joshi, H.; Sharma, U.; Sharma, B.; Aggarwal, D.; Rani, I.; Ramniwas, S.; Gupta, M.; Tuli, H.S. Melittin: A possible regulator of cancer proliferation in preclinical cell culture and animal models. J. Cancer Res. Clin. Oncol. 2023, 149, 17709–17726. [Google Scholar] [CrossRef]
- Uraki, S.; Sugimoto, K.; Shiraki, K.; Tameda, M.; Inagaki, Y.; Ogura, S.; Kasai, C.; Nojiri, K.; Yoneda, M.; Yamamoto, N.; et al. Human β-defensin-3 inhibits migration of colon cancer cells via downregulation of metastasis-associated 1 family, member 2 expression. Int. J. Oncol. 2014, 45, 1059–1064. [Google Scholar] [CrossRef]
- Wang, J.; Cheng, M.; Law, I.K.M.; Ortiz, C.; Sun, M.; Koon, H.W. Cathelicidin suppresses colon cancer metastasis via a P2RX7-dependent mechanism. Mol. Ther. Oncolytics 2019, 12, 195–203. [Google Scholar] [CrossRef]
- Jafari, A.; Babajani, A.; Sarrami Forooshani, R.; Yazdani, M.; Rezaei-Tavirani, M. Clinical Applications and Anticancer Effects of Antimicrobial Peptides: From Bench to Bedside. Front. Oncol. 2022, 12, 819563. [Google Scholar] [CrossRef] [PubMed]
- Paredes-Gamero, E.J.; Martins, M.N.; Cappabianco, F.A.; Ide, J.S.; Miranda, A. Characterization of dual effects induced by antimicrobial peptides: Regulated cell death or membrane disruption. Biochim. Biophys. Acta 2012, 1820, 1062–1072. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, T.; Guo, Y.; Bi, C.; Liu, M.; Wang, G. Biological impact and therapeutic implication of tumor-associated macrophages in hepatocellular carcinoma. Cell Death Dis. 2024, 15, 498. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Han, G.; Zhang, Y.; Zhang, L.; Li, Z.; Wang, Q.; Chen, Z.; Wang, X.; Wu, J. M2 macrophage-secreted exosomes promote metastasis and increase vascular permeability in hepatocellular carcinoma. Cell Commun. Signal. 2023, 21, 299. [Google Scholar] [CrossRef]
- Cheng, K.; Cai, N.; Zhu, J.; Yang, X.; Liang, H.; Zhang, W. Tumor-associated macrophages in liver cancer: From mechanisms to therapy. Cancer Commun. 2022, 42, 1112–1140. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, S.; Hu, M.; Liu, Z.; Yu, P.; Li, C.; Zhang, X. Antibacterial and Anti-Inflammatory Properties of Peptide KN-17. Microorganisms 2022, 10, 2114. [Google Scholar] [CrossRef]
- Sun, Y.; Li, H.; Duan, X.; Ma, X.; Liu, C.; Shang, D. Chensinin-1b Alleviates DSS-Induced Inflammatory Bowel Disease by Inducing Macrophage Switching from the M1 to the M2 Phenotype. Biomedicines 2024, 12, 345. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, X.; Zhang, H.; Zhu, Y.; Xu, K.; Yang, Y.; Xie, W.; Duan, W.; Chen, Q.; Chen, Y. Myristoyl-CM4 Exhibits Direct Anticancer Activity and Immune Modulation in Hepatocellular Carcinoma: Evidence from In Vitro and Mouse Model Studies. Int. J. Mol. Sci. 2025, 26, 3829. https://doi.org/10.3390/ijms26083829
Yuan X, Zhang H, Zhu Y, Xu K, Yang Y, Xie W, Duan W, Chen Q, Chen Y. Myristoyl-CM4 Exhibits Direct Anticancer Activity and Immune Modulation in Hepatocellular Carcinoma: Evidence from In Vitro and Mouse Model Studies. International Journal of Molecular Sciences. 2025; 26(8):3829. https://doi.org/10.3390/ijms26083829
Chicago/Turabian StyleYuan, Xueli, Huidan Zhang, Yiqiang Zhu, Ke Xu, Yaxin Yang, Wenjing Xie, Wenliang Duan, Qin Chen, and Yuqing Chen. 2025. "Myristoyl-CM4 Exhibits Direct Anticancer Activity and Immune Modulation in Hepatocellular Carcinoma: Evidence from In Vitro and Mouse Model Studies" International Journal of Molecular Sciences 26, no. 8: 3829. https://doi.org/10.3390/ijms26083829
APA StyleYuan, X., Zhang, H., Zhu, Y., Xu, K., Yang, Y., Xie, W., Duan, W., Chen, Q., & Chen, Y. (2025). Myristoyl-CM4 Exhibits Direct Anticancer Activity and Immune Modulation in Hepatocellular Carcinoma: Evidence from In Vitro and Mouse Model Studies. International Journal of Molecular Sciences, 26(8), 3829. https://doi.org/10.3390/ijms26083829