
4-Substituted Pyridine-3-Sulfonamides as Carbonic Anhydrase Inhibitors Modified by Click Tailing: Synthesis, Activity, and Docking Studies
 , ,
, ,  ,
,  , , and
, , and 
Abstract

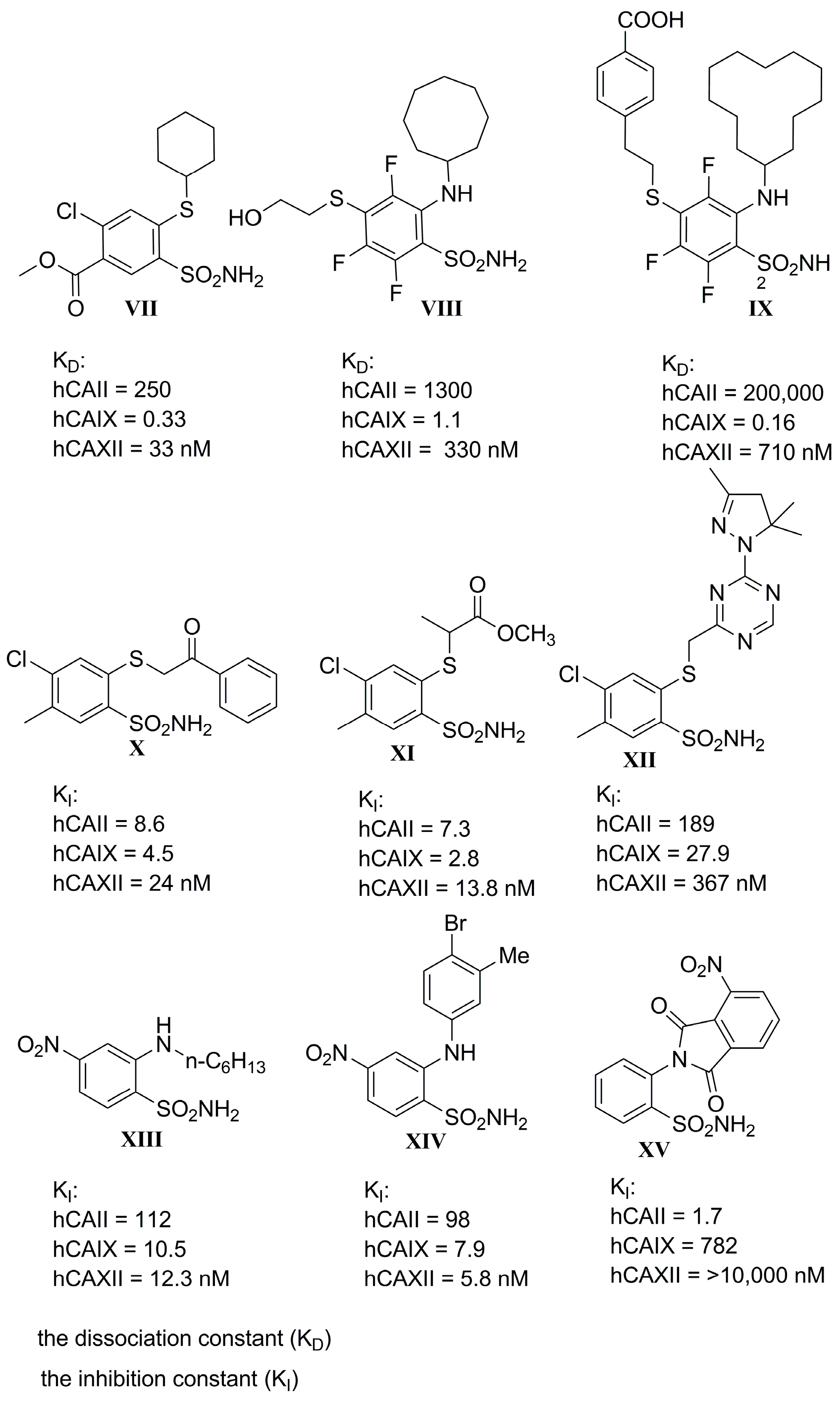
1. Introduction
2. Results and Discussion
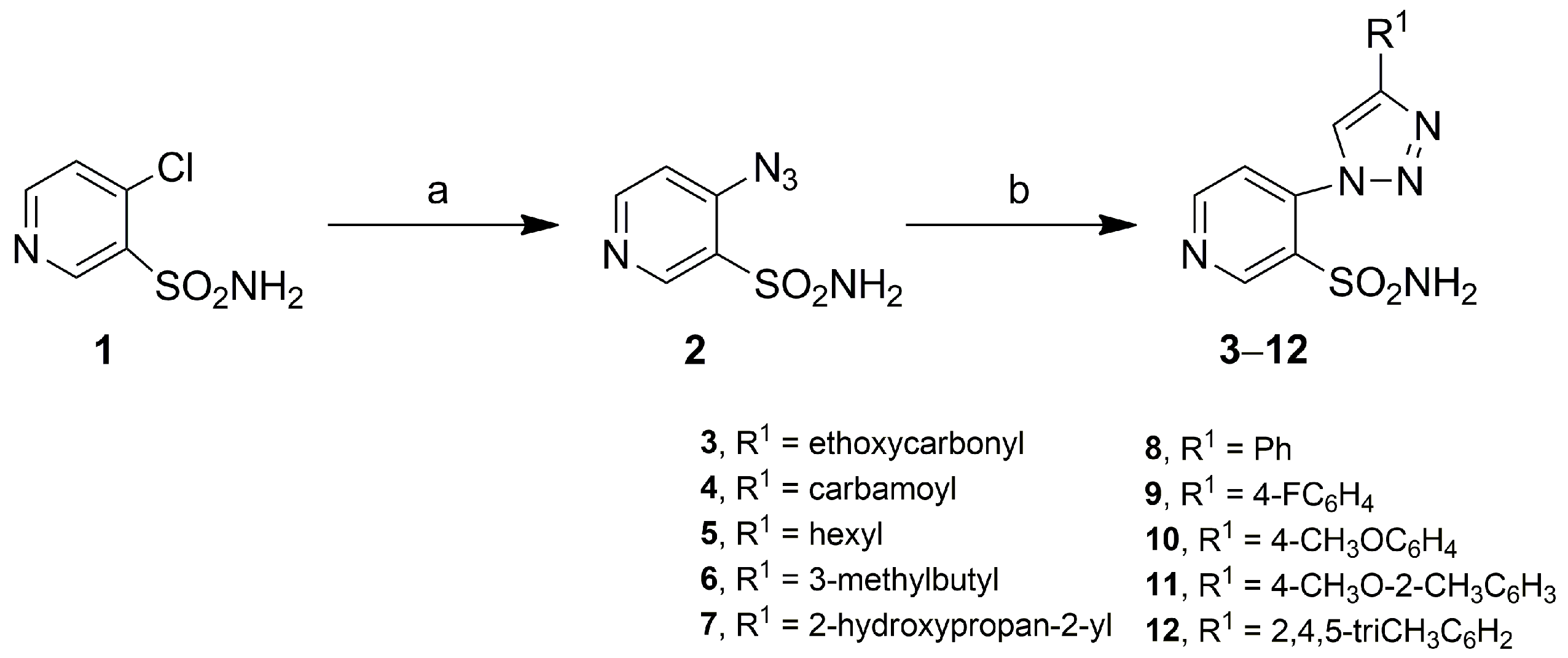
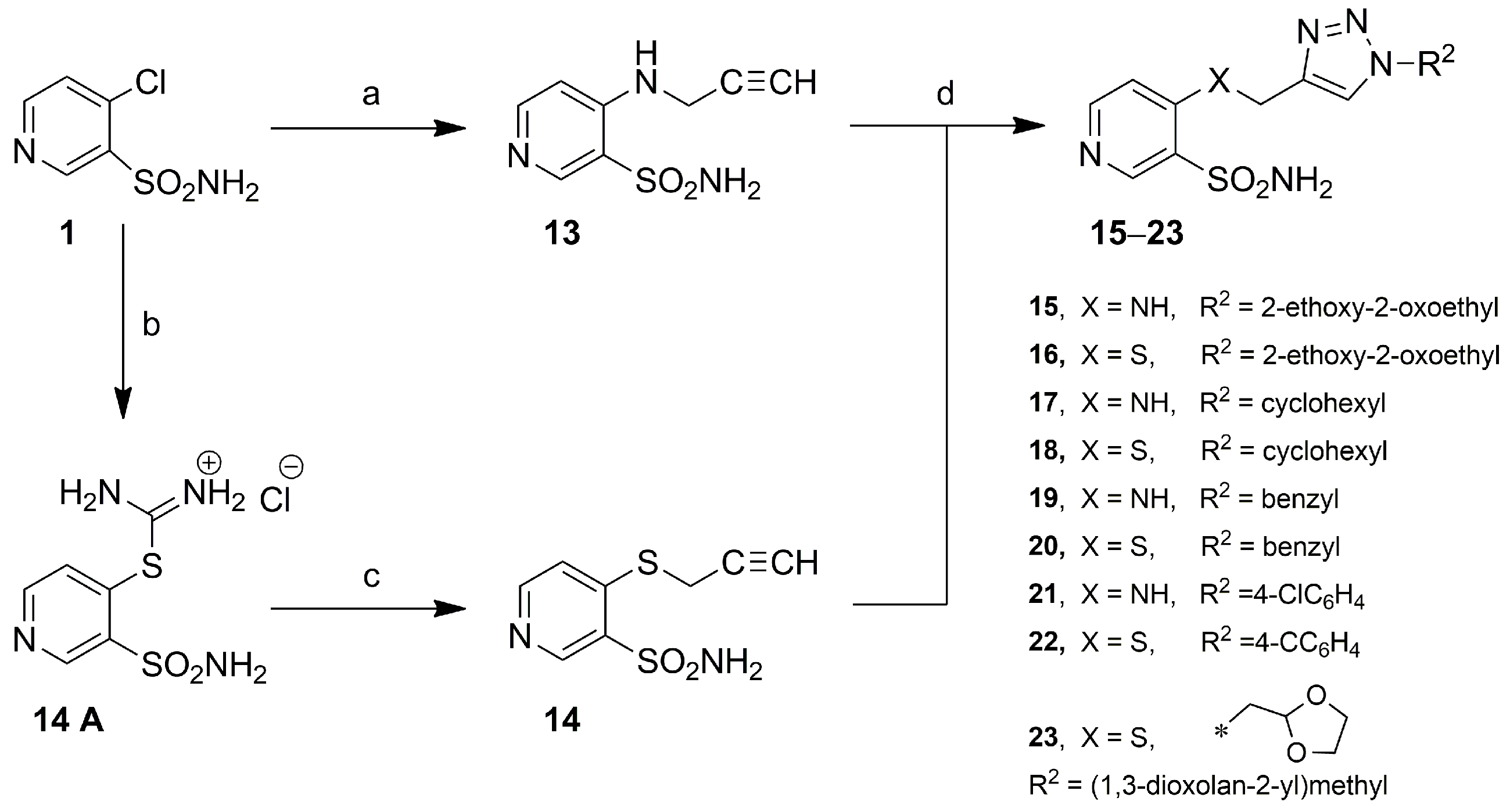
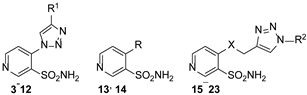
2.1. Chemistry
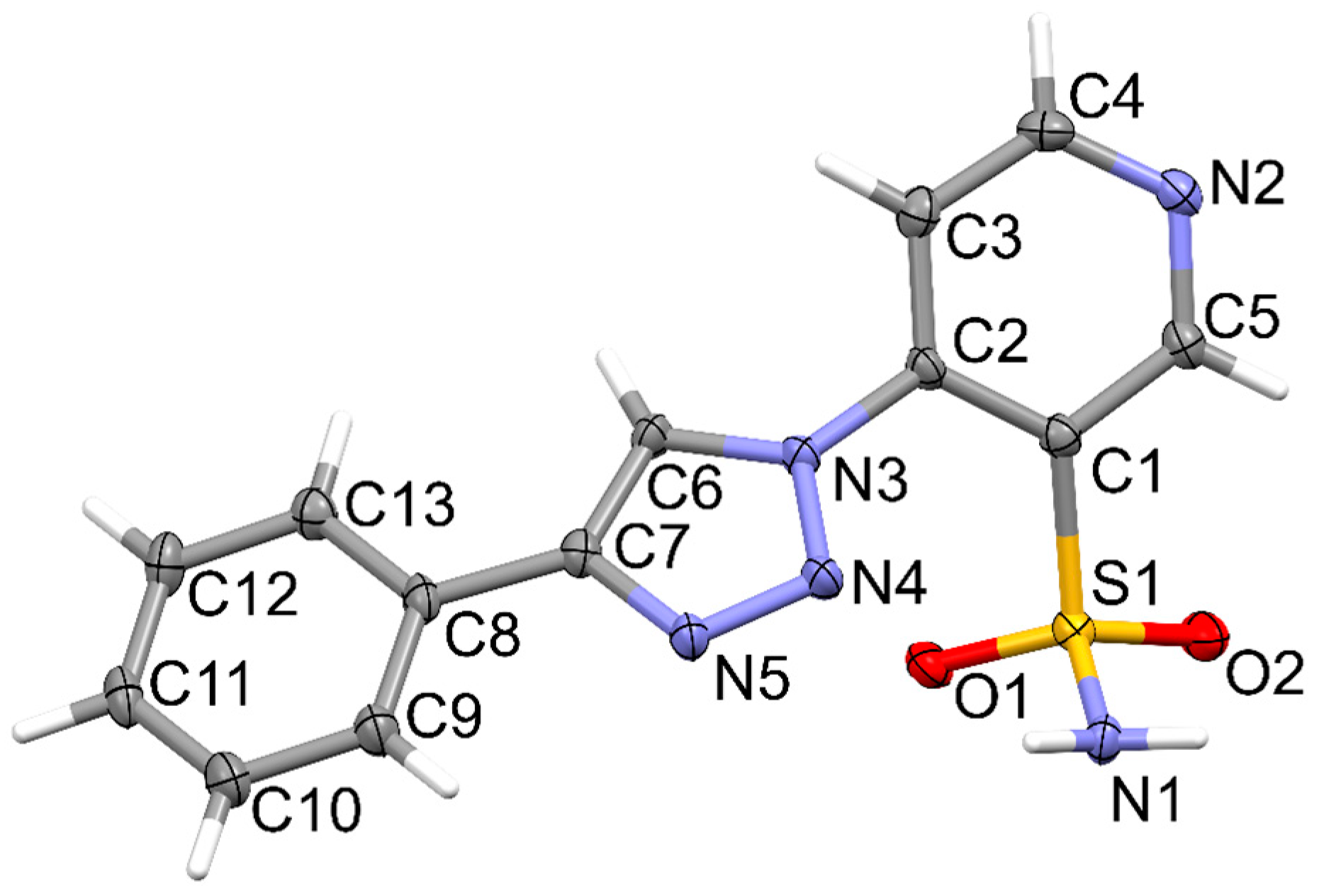
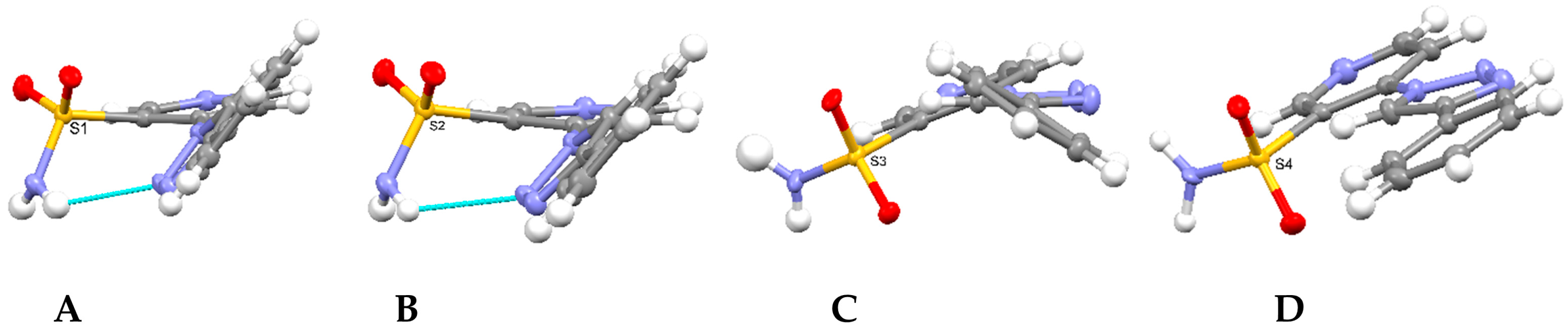
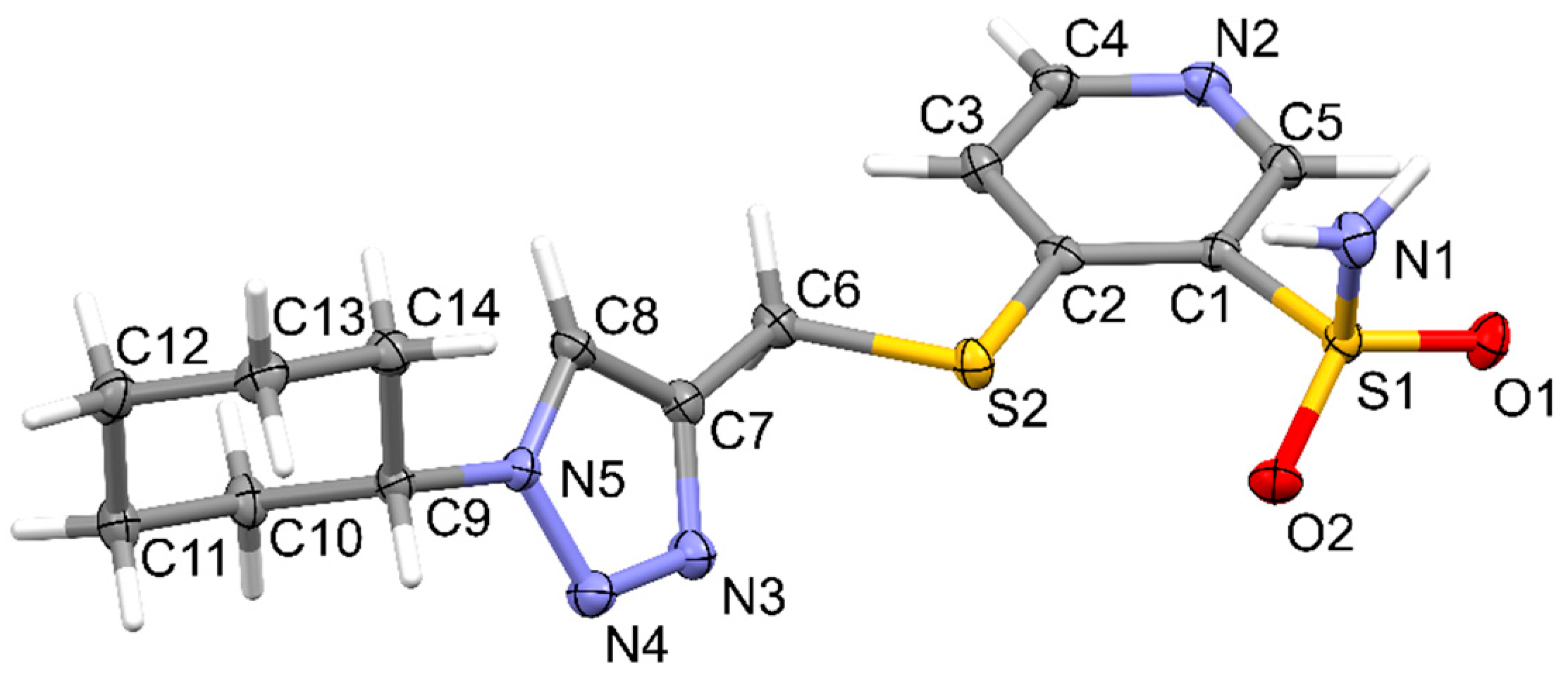
2.2. Crystallographic Studies
2.3. Carbonic Anhydrase Inhibition
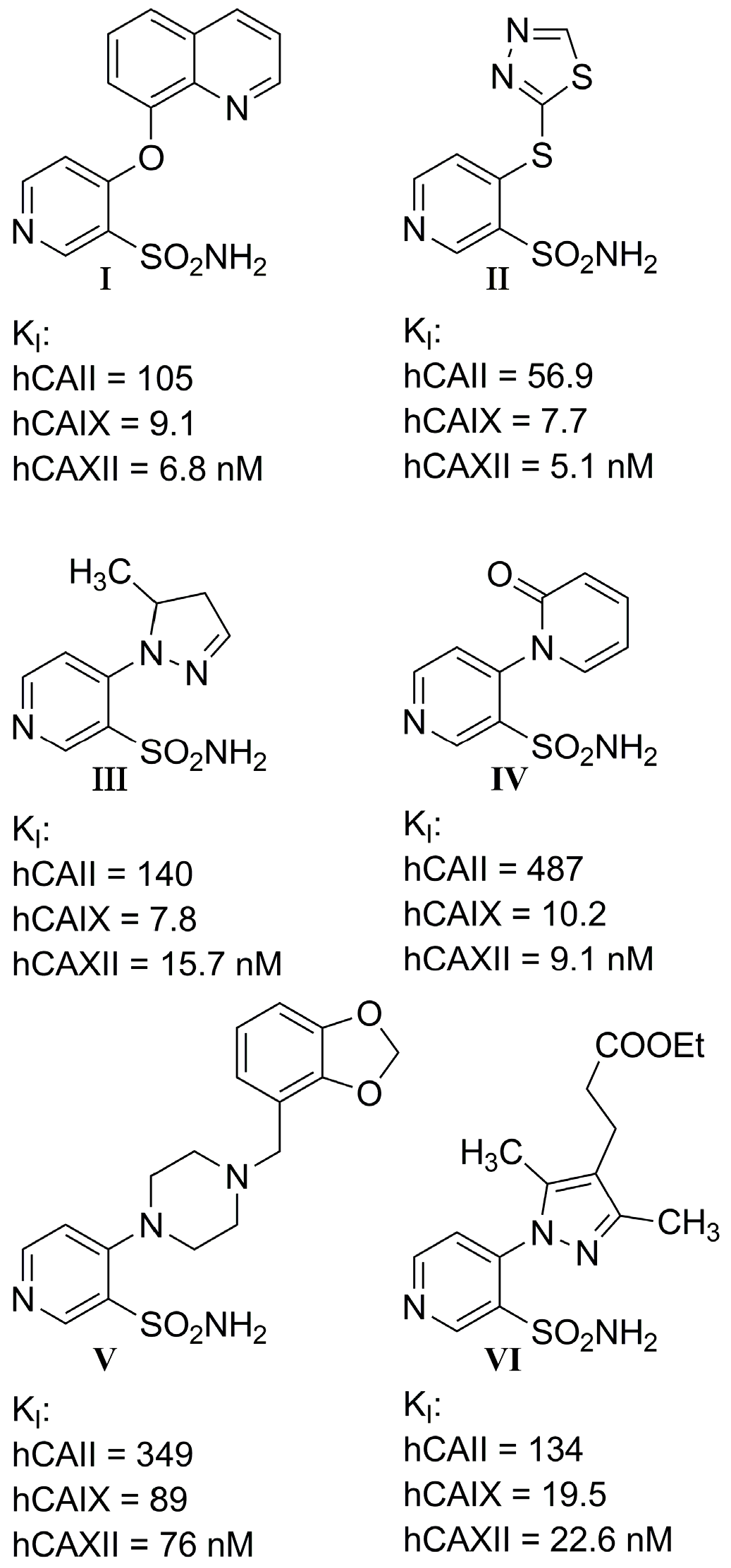
- I.
- Most compounds exhibited a similar or only slightly varied (compounds 4 and 7) rank order of potency (KI-CAXII < KI-CAIX < KI-CAII), except for pairs 5–6 and 15–16. Moreover, a statistically significant correlation can be observed between the activity against hCA II and hCA IX, with a Pearson correlation coefficient of 0.92.
- II.
- The inhibitory activity against the ubiquitous and fast-reacting isoform hCA II showed the greatest variability, ranging from 271.5 nM for compound 5 to over 10,000 nM for compounds 12, 21, and 22. Among the derivatives 3–12, in which the triazole ring is directly linked to pyridine, those containing a phenyl R substituent 8–12 were markedly less active, with potency further diminishing as the bulk and number of substituents increased. This trend was also observed across other tested isoforms. Conversely, aliphatic lipophilic substituents, such as n-hexyl (5) and 3-methylbutan-1-yl (6), showed the highest activity against hCA II in this series. A similar preference for aliphatic lipophilic substituents was observed in the 15–23 series, where compounds 17, 18, and 23 demonstrated KI values of 419 nM, 505 nM, and 709 nM, respectively.
- III.
- The activity against hCA IX ranged from 137.5 nM for compound 4 to 8154 nM for compound 22. In the 3–12 series, contrary to the trend observed for hCA II, the presence of aliphatic R1 substituents in compounds 5 and 6 significantly reduced activity compared to compounds 3, 4, and 7. All compounds in the 15–23 series were more active against hCA IX than hCA II, with the highest activity observed for compounds 17 and 18 (R2 = cyclohexyl) and compound 23 (R2 = methyl-1,3-dioxolane).
- IV.
- The most sensitive isoform, hCA XII, was inhibited at concentrations as low as 91 nM by compounds 6, 18, and 23, while compound 16, the least active, exhibited a KI of 4284 nM. Similar to hCA II, a positive impact of aliphatic R1 substituents in compounds 5 and 6 was observed, with these compounds displaying greater activity than compound 4, which has an amide R1 substituent, and compound 7, which has an alcohol R1 substituent.
- V.
- For the derivatives 15–22, the nature of either the thiomethyl (-S-CH2-) or aminomethyl (-NH-CH2-) linker did not appear to critically influence activity, particularly against hCA II, as pairs of compounds with the same R2 exhibited similar activity profiles (refer to the heatmap in the table). For other isoforms, the effect of swapping sulfur and nitrogen is challenging to predict. For instance, in hCA XII, changing X from N to S can either halve the KI (compounds 17–18 and 19–20) or double it (compounds 15–16 and 21–22), whereas in hCA IX, the same substitution yields the opposite effect (pairs 17–18 and 19–20). The modification of the R2 substituent had a more pronounced effect.
- VI.
- In terms of selectivity toward tumor-associated isoforms IX and XII, it is noteworthy that compounds 4, 7, 15, 16, and 23 exhibited moderate (3 to 5.9-fold) selectivity for isoform hCA IX over isoform hCA II. The new compounds demonstrated significantly higher selectivity for hCA XII, with compounds 20, 8, 10, and 9 exhibiting KI values 9.1 to 18.7-times lower than those for hCA II.
- VII.
- Selectivity between the two tumor-associated isoforms is also significant, with compounds 6 (23.3-fold), 5 (18.2-fold), 20 (10.5-fold), and 8 (9.3-fold) favoring hCA XII, while compounds 16 (8.0-fold), 4 (4.4-fold), and 15 (4.0-fold) showed preference for hCA IX.
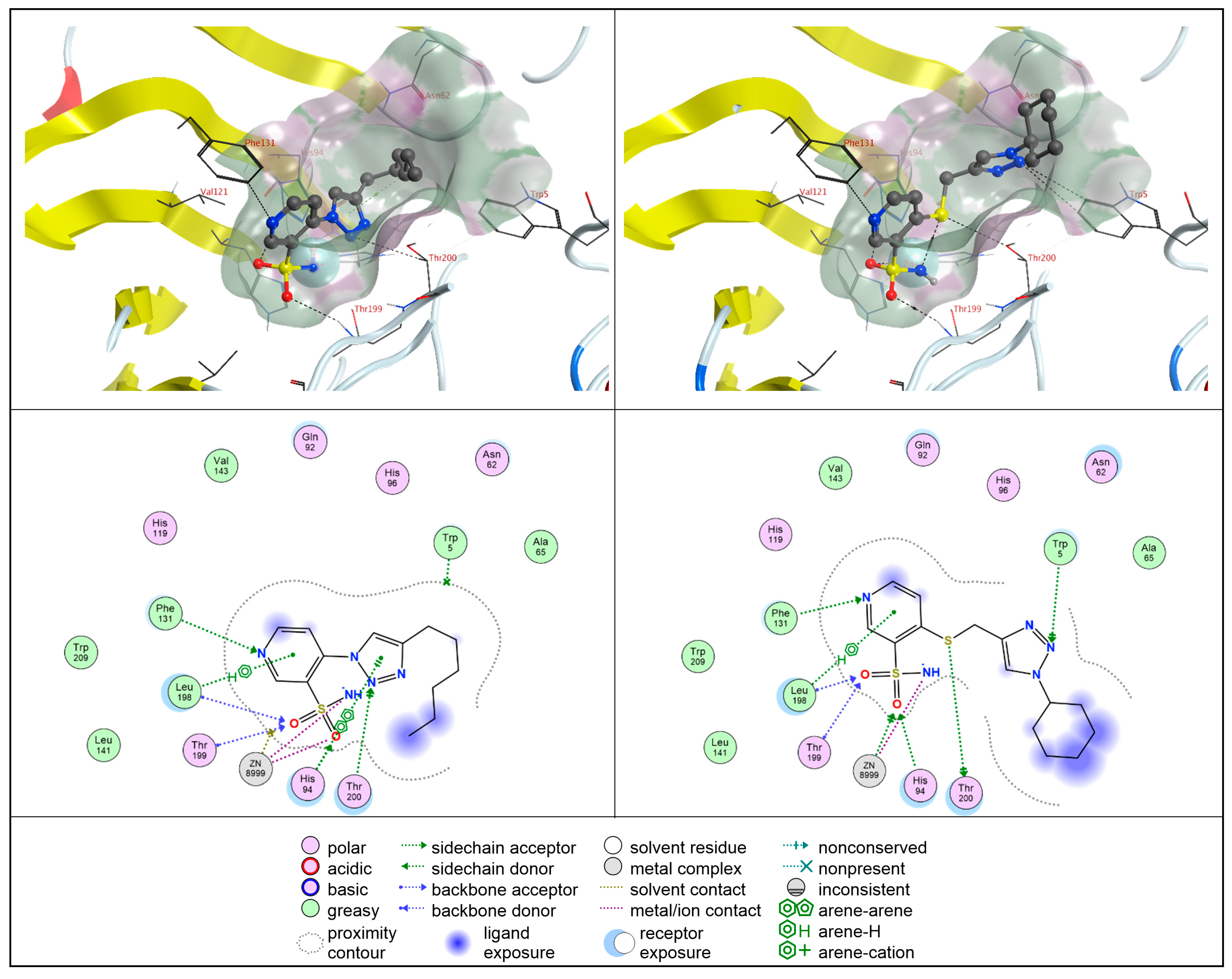
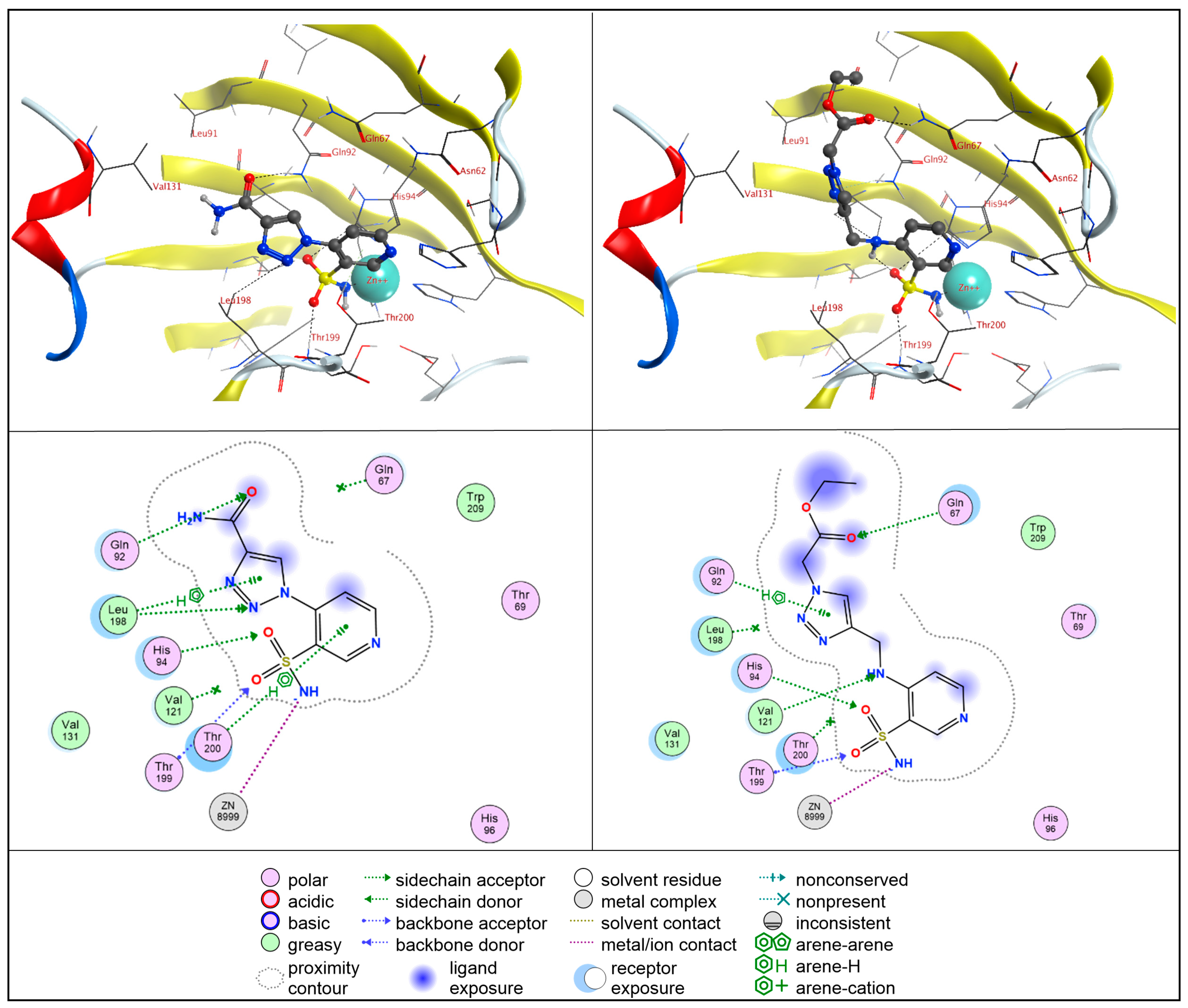
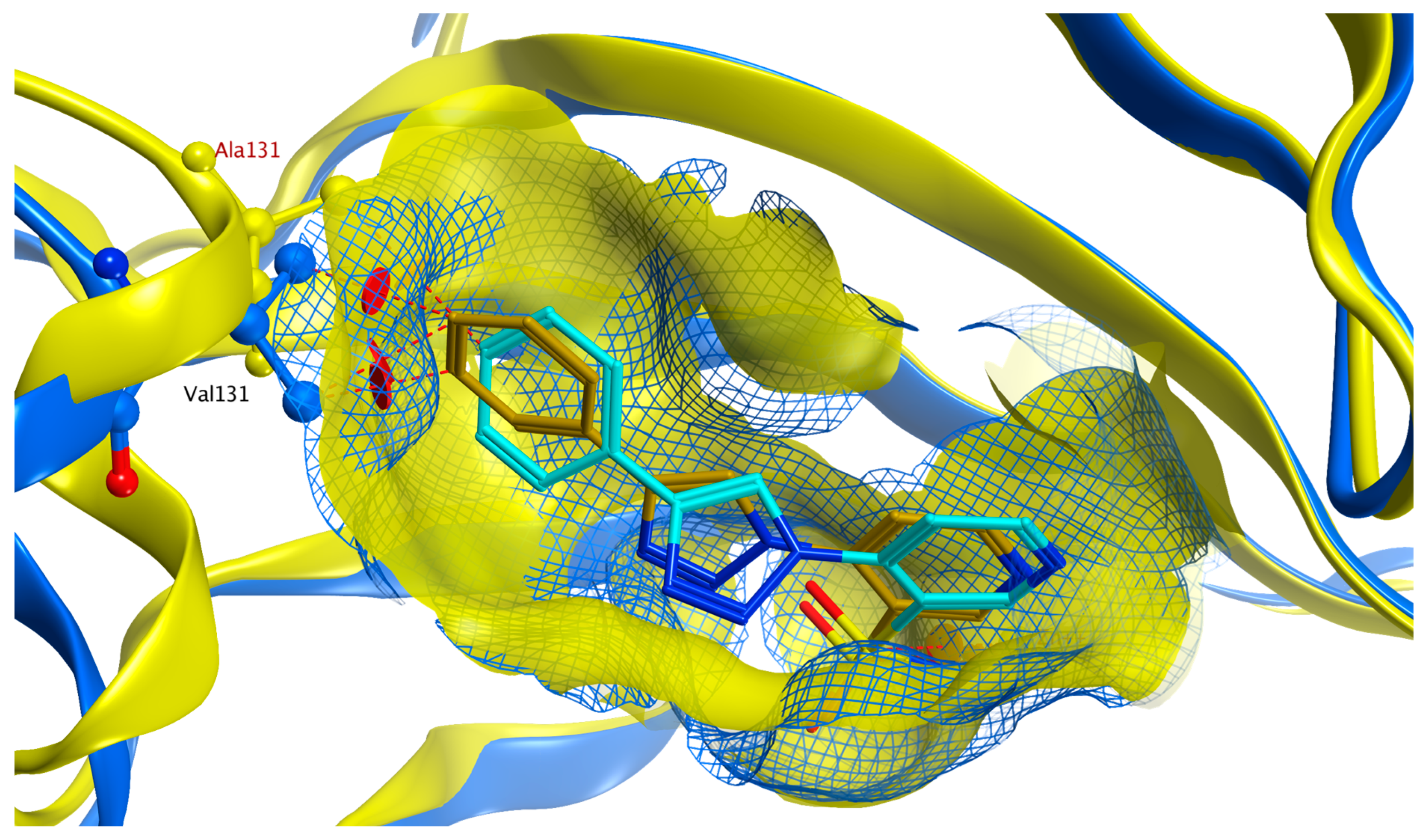
2.4. Molecular Docking Studies
2.4.1. hCA II
2.4.2. hCA IX
2.4.3. hCA XII
2.4.4. Antiproliferative Screening
3. Materials and Methods
3.1. Carbonic Anhydrase Inhibition Assay
3.2. Cell Culture and Viability Assay
3.3. X-Ray Structure Determination
3.4. Synthesis
3.4.1. General Method for Obtaining 4-(4-Methyl-1H-1,2,3-triazol-1-yl)pyridine-3-Sulfonamides 3–12
Ethyl 1-(3-Sulfamoylpyridin-4-yl)-1H-1,2,3-triazole-4-carboxylate (3)
1-(3-Sulfamoylpyridin-4-yl)-1H-1,2,3-triazole-4-carboxamide (4)
4-(4-Hexyl-1H-1,2,3-triazol-1-yl)pyridine-3-sulfonamide (5)
4-(4-(3-Methylbutyl)-1H-1,2,3-triazol-1-yl)pyridine-3-sulfonamide (6)
4-(4-(2-Hydroxypropan-2-yl)-1H-1,2,3-triazol-1-yl)pyridine-3-sulfonamide (7)
4-(4-Phenyl-1H-1,2,3-triazol-1-yl)pyridine-3-sulfonamide (8)
4-(4-(4-Fluorophenyl)-1H-1,2,3-triazol-1-yl)pyridine-3-sulfonamide (9)
4-(4-(4-Methoxyphenyl)-1H-1,2,3-triazol-1-yl)pyridine-3-sulfonamide (10)
4-(4-(4-Methoxy-2-methylphenyl)-1H-1,2,3-triazol-1-yl)pyridine-3-sulfonamide (11)
4-(4-(2,4,5-Trimethylphenyl)-1H-1,2,3-triazol-1-yl)pyridine-3-sulfonamide (12)
3.4.2. 4-((Prop-2-yn-1-yl)amino)pyridine-3-sulfonamide (13)
3.4.3. General Method for Obtaining 4-(((1-Substituted-1H-1,2,3-triazol-4-yl)methyl)amino/thio)pyridine-3-sulfonamides 15–23
Ethyl 2-(4-(((3-sulfamoylpyridin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)acetate (15)
Ethyl 2-(4-(((3-Sulfamoylpyridin-4-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)acetate (16)
4-(((1-Cyclohexyl-1H-1,2,3-triazol-4-yl)methyl)amino)pyridine-3-sulfonamide (17)
4-(((1-Cyclohexyl-1H-1,2,3-triazol-4-yl)methyl)thio)pyridine-3-sulfonamide (18)
4-(((1-Benzyl-1H-1,2,3-triazol-4-yl)methyl)amino)pyridine-3-sulfonamide (19)
4-(((1-Benzyl-1H-1,2,3-triazol-4-yl)methyl)thio)pyridine-3-sulfonamide (20)
4-(((1-(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)amino)pyridine-3-sulfonamide (21)
4-(((1-(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)thio)pyridine-3-sulfonamide (22)
4-(((1-((1,3-Dioxolan-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)thio)pyridine-3-sulfonamide (23)
3.5. Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mishra, C.B.; Tiwari, M.; Supuran, C.T. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: Where are we today? Med. Res. Rev. 2020, 40, 2485–2565. [Google Scholar] [CrossRef]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Mojzych, M.; Marciniak, B.; Drozda, R.; Kontek, R. Targeting carbonic anhydrase IX and XII isoforms with small molecule inhibitors and monoclonal antibodies. J. Enzym. Inhib. Med. Chem. 2022, 37, 1278–1298. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.C.; Chia, S.; Bedard, P.L.; Chu, Q.; Lyle, M.; Tang, L.; Singh, M.; Zhang, Z.; Supuran, C.T.; Renouf, D.J.; et al. A Phase 1 Study of SLC-0111, a Novel Inhibitor of Carbonic Anhydrase IX, in Patients with Advanced Solid Tumors. Am. J. Clin. Oncol. Cancer Clin. Trials 2020, 43, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, S.; Malkova, A.; Sharonova, T.; Sharoyko, V.; Bunev, A.; Supuran, C.T.; Krasavin, M. Carbonic anhydrase IX inhibitors as candidates for combination therapy of solid tumors. Int. J. Mol. Sci. 2021, 22, 13405. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.; Meehan, J.; Gray, M.; Kunkler, I.H.; Langdon, S.P.; Argyle, D.J. Carbonic anhydrase IX (CAIX), cancer, and radiation responsiveness. Metabolites 2018, 8, 13. [Google Scholar] [CrossRef]
- Clément, S.; Richeter, S.; Winum, J.Y. Targeting Tumor-Associated Carbonic Anhydrases in Photothermal Therapy. ChemMedChem 2025, e202400893. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.C.; Dedhar, S. Co-vulnerabilities of inhibiting carbonic anhydrase IX in ferroptosis-mediated tumor cell death. Front. Mol. Biosci. 2023, 10, 1327310. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, L.; Chew, S.H.; Hirayama, T.; Sekido, Y.; Toyokuni, S. Carbonic anhydrase 9 confers resistance to ferroptosis/apoptosis in malignant mesothelioma under hypoxia. Redox Biol. 2019, 26, 101297. [Google Scholar] [CrossRef]
- Yang, C.; Sun, X.; Li, Z.; Cheng, Y.; Lei, Y.; Lu, L.; Liu, X.; Zhuang, X.; Wang, T.; He, X. The effect of benzenesulfonamide’s side chains on their human carbonic anhydrase I/II inhibitory activities. J. Mol. Struct. 2022, 1250, 131927. [Google Scholar] [CrossRef]
- Ahmed, R.F.; Mahmoud, W.R.; Abdelgawad, N.M.; Fouad, M.A.; Said, M.F. Exploring novel anticancer pyrazole benzenesulfonamides featuring tail approach strategy as carbonic anhydrase inhibitors. Eur. J. Med. Chem. 2023, 261, 115805. [Google Scholar] [CrossRef]
- Hou, Z.; Lin, B.; Bao, Y.; Yan, H.N.; Zhang, M.; Chang, X.W.; Zhang, X.X.; Wang, Z.J.; Wei, G.F.; Cheng, M.S.; et al. Dual-tail approach to discovery of novel carbonic anhydrase IX inhibitors by simultaneously matching the hydrophobic and hydrophilic halves of the active site. Eur. J. Med. Chem. 2017, 132, 1–10. [Google Scholar] [CrossRef] [PubMed]
- El-Hazek, R.M.M.; Zaher, N.H.; Emam, H.E.S.; El-Gazzar, M.G.; Khalil, A. Pyrazole-sulfonamide scaffold featuring dual-tail strategy as apoptosis inducers in colon cancer. Sci. Rep. 2023, 13, 5782. [Google Scholar] [CrossRef] [PubMed]
- Zakšauskas, A.; Čapkauskaitė, E.; Jezepčikas, L.; Linkuvienė, V.; Kišonaitė, M.; Smirnov, A.; Manakova, E.; Gražulis, S.; Matulis, D. Design of two-tail compounds with rotationally fixed benzenesulfonamide ring as inhibitors of carbonic anhydrases. Eur. J. Med. Chem. 2018, 156, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Borras, J.; Scozzafava, A.; Menabuoni, L.; Mincione, F.; Briganti, F.; Supuran, C.T. Carbonic Anhydrase Inhibitors: Synthesis of Water-Soluble, Topically Eective Intraocular Pressure Lowering Aromatic/Heterocyclic Sulfonamides Containing 8-Quinoline-sulfonyl Moieties: Is the Tail More Important than the Ring? J. Med. Chem. 1999, 7, 2397–2406. [Google Scholar]
- Sławiński, J.; Szafrański, K.; Vullo, D.; Supuran, C.T. Carbonic anhydrase inhibitors. Synthesis of heterocyclic 4-substituted pyridine-3-sulfonamide derivatives and their inhibition of the human cytosolic isozymes I and II and transmembrane tumor-associated isozymes IX and XII. Eur. J. Med. Chem. 2013, 69, 701–710. [Google Scholar] [CrossRef]
- Brzozowski, Z.; Sławiński, J.; Saczewski, F.; Innocenti, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Synthesis and inhibition of the human cytosolic isozymes I and II and transmembrane isozymes IX, XII (cancer-associated) and XIV with 4-substituted 3-pyridinesulfonamides. Eur. J. Med. Chem. 2010, 45, 2396–2404. [Google Scholar] [CrossRef]
- Brzozowski, Z.; Sławiński, J.; Gdaniec, M.; Innocenti, A.; Supuran, C.T. Carbonic anhydrase inhibitors. Synthesis, molecular structures, and inhibition of the human cytosolic isozymes i and II and transmembrane isozymes IX, XII (cancer-associated) and XIV with novel 3-pyridinesulfonamide derivatives. Eur. J. Med. Chem. 2011, 46, 4403–4410. [Google Scholar] [CrossRef]
- Zakšauskas, A.; Čapkauskaitė, E.; Paketurytė-Latvė, V.; Smirnov, A.; Leitans, J.; Kazaks, A.; Dvinskis, E.; Stančaitis, L.; Mickevičiūtė, A.; Jachno, J.; et al. Methyl 2-halo-4-substituted-5-sulfamoyl-benzoates as high affinity and selective inhibitors of carbonic anhydrase ix. Int. J. Mol. Sci. 2022, 23, 130. [Google Scholar] [CrossRef]
- Kazokaite, J.; Niemans, R.; Dudutiene, V.; Becker, H.M.; Leitans, J.; Zubriene, A.; Baranauskiene, L.; Gondi, G.; Zeidler, R.; Matuliene, J.; et al. Novel fluorinated carbonic anhydrase IX inhibitors reduce hypoxia-induced acidification and clonogenic survival of cancer cells. Oncotarget 2018, 9, 26800–26816. [Google Scholar] [CrossRef]
- Saczewski, F.; Innocenti, A.; Sławiński, J.; Kornicka, A.; Brzozowski, Z.; Pomarnacka, E.; Scozzafava, A.; Temperini, C.; Supuran, C.T. Carbonic anhydrase inhibitors: Inhibition of human cytosolic isozymes I and II and tumor-associated isozymes IX and XII with S-substituted 4-chloro-2-mercapto-5-methyl-benzenesulfonamides. Bioorg. Med. Chem. 2008, 16, 3933–3940. [Google Scholar] [CrossRef]
- D’Ambrosio, K.; Vitale, R.M.; Dogné, J.M.; Masereel, B.; Innocenti, A.; Scozzafava, A.; De Simone, G.; Supuran, C.T. Carbonic anhydrase inhibitors: Bioreductive nitro-containing sulfonamides with selectivity for targeting the tumor associated isoforms IX and XII. J. Med. Chem. 2008, 51, 3230–3237. [Google Scholar] [CrossRef] [PubMed]
- Sethi, K.K.; Verma, S.M.; Tanç, M.; Purper, G.; Calafato, G.; Carta, F.; Supuran, C.T. Carbonic anhydrase inhibitors: Synthesis and inhibition of the human carbonic anhydrase isoforms I, II, IX and XII with benzene sulfonamides incorporating 4- and 3-nitrophthalimide moieties. Bioorg. Med. Chem. 2014, 22, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- Kakakhan, C.; Türkeş, C.; Güleç, Ö.; Demir, Y.; Arslan, M.; Özkemahlı, G.; Beydemir, Ş. Exploration of 1,2,3-triazole linked benzenesulfonamide derivatives as isoform selective inhibitors of human carbonic anhydrase. Bioorg. Med. Chem. 2023, 77, 117111. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Cheng, X.; An, X.W.R.; Xu, H.; Guo, M.; Li, C.; Wang, Y.; Hou, Z.; Guo, C. Design, synthesis and biological evaluation of novel carbohydrate-based sulfonamide derivatives as antitumor agents. Bioorg. Chem. 2020, 104, 104237. [Google Scholar] [CrossRef]
- Cuffaro, D.; Nuti, E.; Rossello, A. An overview of carbohydrate-based carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem. 2020, 35, 1906–1922. [Google Scholar] [CrossRef]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple binding modes of inhibitors to carbonic anhydrases: How to design specific drugs targeting 15 different isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef]
- Whittington, D.A.; Waheed, A.; Ulmasov, B.; Shah, G.N.; Grubb, J.H.; Sly, W.S.; Christianson, D.W. Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc. Natl. Acad. Sci. USA 2001, 98, 9545–9550. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE). 2022.02 Chemical Computing Group ULC; Molecular Operating Environment (MOE): Montreal, QC, Canada, 2023. [Google Scholar]
- Dudutiene, V.; Matuliene, J.; Smirnov, A.; Timm, D.D.; Zubriene, A.; Baranauskiene, L.; Morkunaite, V.; Smirnoviene, J.; Michailoviene, V.; Juozapaitiene, V.; et al. Discovery and characterization of novel selective inhibitors of carbonic anhydrase IX. J. Med. Chem. 2014, 57, 9435–9446. [Google Scholar] [CrossRef]
- Zubrienė, A.; Smirnov, A.; Dudutienė, V.; Timm, D.D.; Matulienė, J.; Michailovienė, V.; Zakšauskas, A.; Manakova, E.; Gražulis, S.; Matulis, D. Intrinsic Thermodynamics and Structures of 2,4- and 3,4-Substituted Fluorinated Benzenesulfonamides Binding to Carbonic Anhydrases. Chem. Med. Chem. 2017, 12, 161–176. [Google Scholar] [CrossRef]
- Smirnov, A.; Manakova, E.; Matulis, D. Correlations Between Inhibitor Binding Thermodynamics and Co-crystal Structures with Carbonic Anhydrases. In Carbon. Anhydrase as Drug Target; Springer International Publishing: Cham, Switzerland, 2019; pp. 249–261. [Google Scholar] [CrossRef]
- Khalifah, R.G. The Carbon Dioxide Hydration Activity of Carbonic Anhydrase. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Tran, V.; Chinh; Ruebsam, F.; Murphy, E.; Douglas; Dragovich, P.; Zhou, Y.; Chen, L.; Kucera, D.; Blatter, F.; et al. 5,6-DIHYDRO-1H-PYRIDIN-2-ONE COMPOUNDS. 2008. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2008124450 (accessed on 31 August 2023).
- Phippen, C.B.W.; Goldys, A.M.; McErlean, C.S.P. Stetter reactions of unsaturated 1-acyl-1H-pyrroles. Eur. J. Org. Chem. 2011, 2011, 6957–6964. [Google Scholar] [CrossRef]
- McNulty, J.; Keskar, K. Discovery of a robust and efficient homogeneous silver(I) catalyst for the cycloaddition of azides onto terminal alkynes. Eur. J. Org. Chem. 2012, 2012, 5462–5470. [Google Scholar] [CrossRef]
- Maury, J.; Feray, L.; Bertrand, M.P.; Kapat, A.; Renaud, P. Unexpected conversion of alkyl azides to alkyl iodides and of aryl azides to N-tert-butyl anilines. Tetrahedron 2012, 68, 9606–9611. [Google Scholar] [CrossRef]
- Kolb, H.C.; Cavallaro, C.; Shi, Z.-C.; Gontcharov, A.; Wang, Z.-M.; Richardson, P.F.; Kanamarlapudi, R.C. One Step Synthesis of 1,2,3-triazole Carboxylic Acids. U.S. Patent 20,030,135,050, 17 July 2003. [Google Scholar]
- Singh, M.K.; Tilak, R.; Nath, G.; Awasthi, S.K.; Agarwal, A. Design, synthesis and antimicrobial activity of novel benzothiazole analogs. Eur. J. Med. Chem. 2013, 63, 635–644. [Google Scholar] [CrossRef]
- Ionescu, A.; Cornut, D.; Soriano, S.; Guissart, C.; Van Antwerpen, P.; Jabin, I. Efficient ‘one-pot’ methodology for the synthesis of novel tetrahydro-β-carboline, tetrahydroisoquinoline and tetrahydrothienopyridine derivatives. Tetrahedron Lett. 2013, 54, 6087–6089. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | X | R1/R2 | KI [nM] | SI | |||||
| hCA I | hCA II | hCA IX | hCA XII | II/IX | II/XII | IX/XII | |||
| 3 |  | 10,000 | 557.2 | 283.3 | 184.6 | 1.97 | 3.02 | 1.53 | |
| 4 | 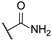 | 10,000 | 811.9 | 137.5 | 599.0 | 5.90 | 1.36 | 0.23 | |
| 5 |  | 10,000 | 271.5 | 1865.0 | 102.4 | 0.15 | 2.65 | 18.21 | |
| 6 |  | 10,000 | 332.4 | 2128.0 | 91.2 | 0.16 | 3.64 | 23.33 | |
| 7 | 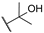 | 10,000 | 602.2 | 178.8 | 474.6 | 3.37 | 1.27 | 0.38 | |
| 8 | 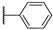 | 10,000 | 4029.0 | 2461.0 | 265.0 | 1.64 | 15.20 | 9.29 | |
| 9 | 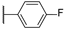 | 10,000 | 3490.0 | 2153.0 | 372.6 | 1.62 | 9.37 | 5.78 | |
| 10 | 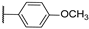 | 10,000 | 5126.0 | 1838.0 | 506.4 | 2.79 | 10.12 | 3.63 | |
| 11 | 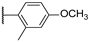 | 10,000 | 7656.0 | 4224.0 | 838.4 | 1.81 | 9.13 | 5.04 | |
| 12 |  | 10,000 | 10,000.0 | 8003.0 | 3047.0 | 1.25 | 3.28 | 2.63 | |
| 15 | NH |  | 10,000 | 1953.0 | 628.9 | 2491.0 | 3.11 | 0.78 | 0.25 |
| 16 | S | 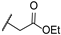 | 10,000 | 2105.0 | 536.5 | 4284.0 | 3.92 | 0.49 | 0.13 |
| 17 | NH | 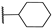 | 10,000 | 419.1 | 285.5 | 172.4 | 1.47 | 2.43 | 1.66 |
| 18 | S | 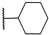 | 10,000 | 505.0 | 199.5 | 90.9 | 2.53 | 5.56 | 2.19 |
| 19 | NH | 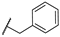 | 10,000 | 6487.0 | 2435.0 | 880.6 | 2.66 | 7.37 | 2.77 |
| 20 | S | 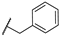 | 10,000 | 7302.0 | 4087.0 | 390.2 | 1.79 | 18.71 | 10.47 |
| 21 | NH | 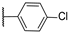 | 10,000 | 10,000.0 | 7711.0 | 1599.0 | 1.30 | 6.25 | 4.82 |
| 22 | S | 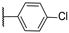 | 10,000 | 10,000.0 | 8154.0 | 2860.0 | 1.23 | 3.50 | 2.85 |
| 23 | S |  | 10,000 | 709.0 | 233.3 | 91.1 | 3.04 | 7.78 | 2.56 |
| AAZ | 250 | 12.0 | 25.0 | 5.7 | 0.48 | 2.1 | 4.39 | ||
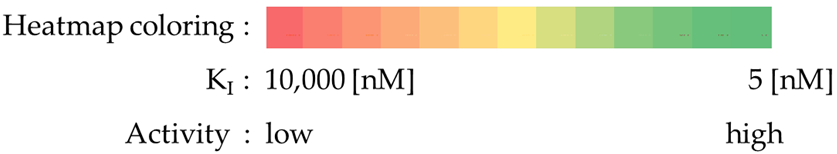 | |||||||||
| IC50 [µM] | ||||
|---|---|---|---|---|
| Compound | HCT-116 | MCF-7 | HeLa | HaCaT |
| 3 | 117 | 120 | 136 | n.t. |
| 4 | 123 | 91 | 148 | n.t. |
| 5 | 65 | 66 | 76 | 105 |
| 6 | 81 | 84 | 90 | n.t. |
| 7 | 120 | 255 | 140 | n.t. |
| 8 | 130 | 300 | 275 | n.t. |
| 9 | 63 | 78 | 71 | n.t. |
| 10 | 83 | 135 | 76 | n.t. |
| 11 | 53 | 56 | 178 | 190 |
| 12 | 17 | 15 | 22 | 45 |
| 15 | 100 | 155 | 115 | n.t. |
| 16 | 230 | 200 | 250 | n.t. |
| 17 | 48 | 45 | 80 | 90 |
| 18 | 77 | * | 82 | 150 |
| 19 | 180 | 105 | 112 | n.t. |
| 20 | 260 | 240 | 245 | n.t. |
| 21 | 66 | 64 | 120 | 125 |
| 22 | 28 | 19 | 39 | n.t. |
| 23 | * | 270 | 460 | n.t. |
| Cisplatin | 3.8 | 3.1 | 2.2 | |
| Doxorubicin | 0.6 | 0.6 | 0.8 | |
| Tamoxifen | 5 | |||
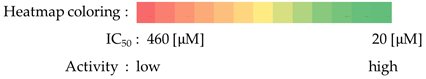 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szafrański, K.; Sławiński, J.; Kawiak, A.; Chojnacki, J.; Kosno, M.; Ammara, A.; Supuran, C.T. 4-Substituted Pyridine-3-Sulfonamides as Carbonic Anhydrase Inhibitors Modified by Click Tailing: Synthesis, Activity, and Docking Studies. Int. J. Mol. Sci. 2025, 26, 3817. https://doi.org/10.3390/ijms26083817
Szafrański K, Sławiński J, Kawiak A, Chojnacki J, Kosno M, Ammara A, Supuran CT. 4-Substituted Pyridine-3-Sulfonamides as Carbonic Anhydrase Inhibitors Modified by Click Tailing: Synthesis, Activity, and Docking Studies. International Journal of Molecular Sciences. 2025; 26(8):3817. https://doi.org/10.3390/ijms26083817
Chicago/Turabian StyleSzafrański, Krzysztof, Jarosław Sławiński, Anna Kawiak, Jarosław Chojnacki, Michał Kosno, Andrea Ammara, and Claudiu T. Supuran. 2025. "4-Substituted Pyridine-3-Sulfonamides as Carbonic Anhydrase Inhibitors Modified by Click Tailing: Synthesis, Activity, and Docking Studies" International Journal of Molecular Sciences 26, no. 8: 3817. https://doi.org/10.3390/ijms26083817
APA StyleSzafrański, K., Sławiński, J., Kawiak, A., Chojnacki, J., Kosno, M., Ammara, A., & Supuran, C. T. (2025). 4-Substituted Pyridine-3-Sulfonamides as Carbonic Anhydrase Inhibitors Modified by Click Tailing: Synthesis, Activity, and Docking Studies. International Journal of Molecular Sciences, 26(8), 3817. https://doi.org/10.3390/ijms26083817